深度解析医学证据,DeepEvidence为你支撑决策
重症肌无力(MG)是一种神经肌肉接头疾病,通常与损害神经肌肉传递的自身抗体(Abs)相关。然而,约 10% 的病例为血清阴性。新兴证据表明,血清阴性重症肌无力(SNMG)可能被遗传性疾病模拟,尤其是先天性肌无力综合征(CMSs),二者治疗方案不同。本研究旨在明确确诊 SNMG 患者中 CMS 的占比。2022 年 8 月至 2024 年 1 月,纳入奥地利 3 家三级神经肌肉疾病诊疗中心确诊为 SNMG 的成年患者(年龄≥18 岁),采用全外显子测序(WES)进行检测。对经全面血清学检测(排除抗(聚集型)乙酰胆碱受体、肌肉特异性激酶、低密度脂蛋白受体相关蛋白 4 及电压门控钙通道抗体)仍为血清阴性的患者进行基因检测。同时,分析与分子诊断可能性相关的临床及人口学因素。
共 50 例 SNMG 患者(35 例 [70%] 为女性)接受基于外显子的基因筛查。疾病起病中位年龄 35 岁(四分位距 24.0~46.0 岁)。经 WES 检测,7 例患者(14%)基因确诊为 CMS(4 例携带 CHRNE 变异,3 例携带 RAPSN 变异)。此外,4 例患者检出临床意义未明的变异,涉及 CACNA1S、DOK7、DPAGT1 及 RAPSN 基因。单因素分析显示,CMS 患者疾病起病年龄更小(p=0.04,r=0.29),但经多重检验校正后,无任何临床或人口学因素与分子诊断显著相关。仅 1 例确诊 CMS 患者报告阳性家族史。6 例 CMS 患者(86%)曾接受免疫调节治疗(n=4)或胸腺切除术(n=4)。4 例接受免疫治疗的 CMS 患者中,3 例报告至少达到部分缓解。
本研究结果证实,相当一部分确诊 SNMG 的患者存在潜在遗传性病因。值得注意的是,对免疫治疗的(主观)反应并不能排除 CMS 分子诊断。综上,为肌无力综合征血清阴性患者提供基因检测可能对治疗决策产生深远影响。
研究背景
重症肌无力(MG)是一种自身免疫介导的神经肌肉接头疾病,表现为劳累性肌无力与易疲劳性。针对突触后抗原(如乙酰胆碱受体 [AChR]、肌肉特异性激酶 [MuSK]、低密度脂蛋白受体相关蛋白 4 [LRP4])的自身抗体(Abs)是重要生物标志物,也是 MG 诊断的核心依据。约 10% 的 MG 病例经常规检测无法检出抗体,归类为血清阴性重症肌无力(SNMG)。
迄今为止,SNMG 的发病机制尚不明确。近期针对 SNMG 患者肌肉组织的病理研究提示补体系统参与其中,证实了疾病的自身免疫性质。尽管如此,部分归类为 SNMG 的患者实则可能存在潜在遗传性疾病,其中最主要的是先天性肌无力综合征(CMSs),其临床表现与自身免疫性 MG 高度重叠。但 CMS 需采用不同治疗方案,根据潜在分子机制,可选用 3,4 - 二氨基吡啶、沙丁胺醇 / 麻黄碱及开放通道阻滞剂等药物。相比之下,乙酰胆碱酯酶抑制剂(AChEIs,如溴吡斯的明)对自身免疫性 MG 及部分 CMS 亚型有效,但对另一些亚型可能有害。
一项回顾性分析显示,约半数成年 CMS 患者在基因检测前被误诊为 SNMG,导致适宜治疗延迟。另一项研究通过对 3 个基因的靶向检测,在 22 例 SNMG 患者队列中发现 1 例 CHRNE 相关 CMS。还有研究通过对 74 例 SNMG 患者进行 RAPSN 与 DOK7 靶向测序,确诊 1 例 RAPSN 相关 CMS。鉴于二者治疗方案截然不同,区分遗传性病因与自身免疫介导的 SNMG 具有重要临床意义。
随着基因组医学的发展,CMS 的分子谱不断完善,目前已明确 35 个致病基因。这提示全外显子测序(WES)等全面分析方法可在 SNMG 队列中识别更多遗传性疾病病例。迄今为止,仅有的一项 WES 研究局限于有阳性家族史的成年起病高度预选小样本病例,确诊率达 28%。但针对更广泛 SNMG 人群、采用全面基因筛查方法的系统性研究仍缺乏。为弥补这一空白,本研究前瞻性对三级转诊中心 50 例确诊 SNMG 的特征明确队列应用 WES,以识别未确诊的 CMS 病例。同时,提供基因确诊病例的表型信息,旨在预测基因诊断的人口学与临床因素。
研究结果
队列人口学与临床特征:
共 50 例 SNMG 患者接受 WES,女性占 70%。疾病起病中位年龄 35 岁(四分位距 [IQR] 24.0~46.0 岁),8% 患者报告 MG 阳性家族史。总计 80% 患者接受过免疫治疗,38% 接受胸腺切除术。病程中任意时间点,47 例(94%)出现眼肌症状,29 例(58%)出现肢体无力,26 例(52%)出现球部症状,16 例(32%)出现呼吸受累。入组时 MG-ADL 评分中位值 4 分(IQR 1.8~8.0)。队列整体及基因确诊 / 未确诊病例的人口学与临床特征总结于表 1。
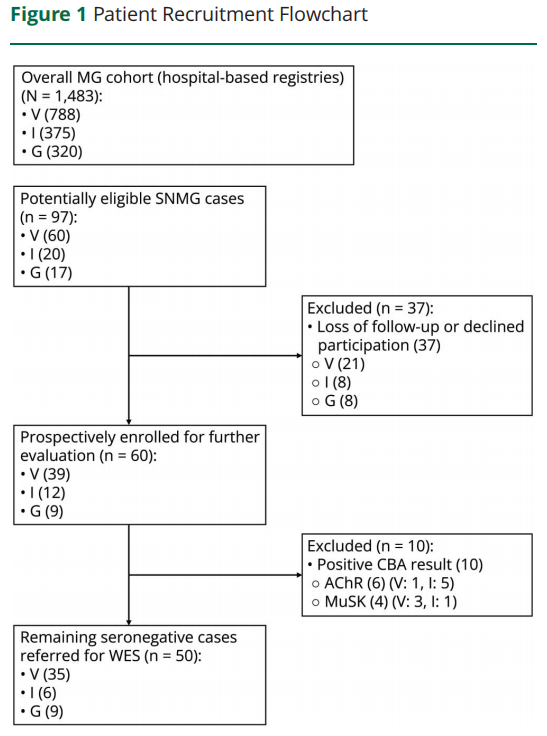
图1
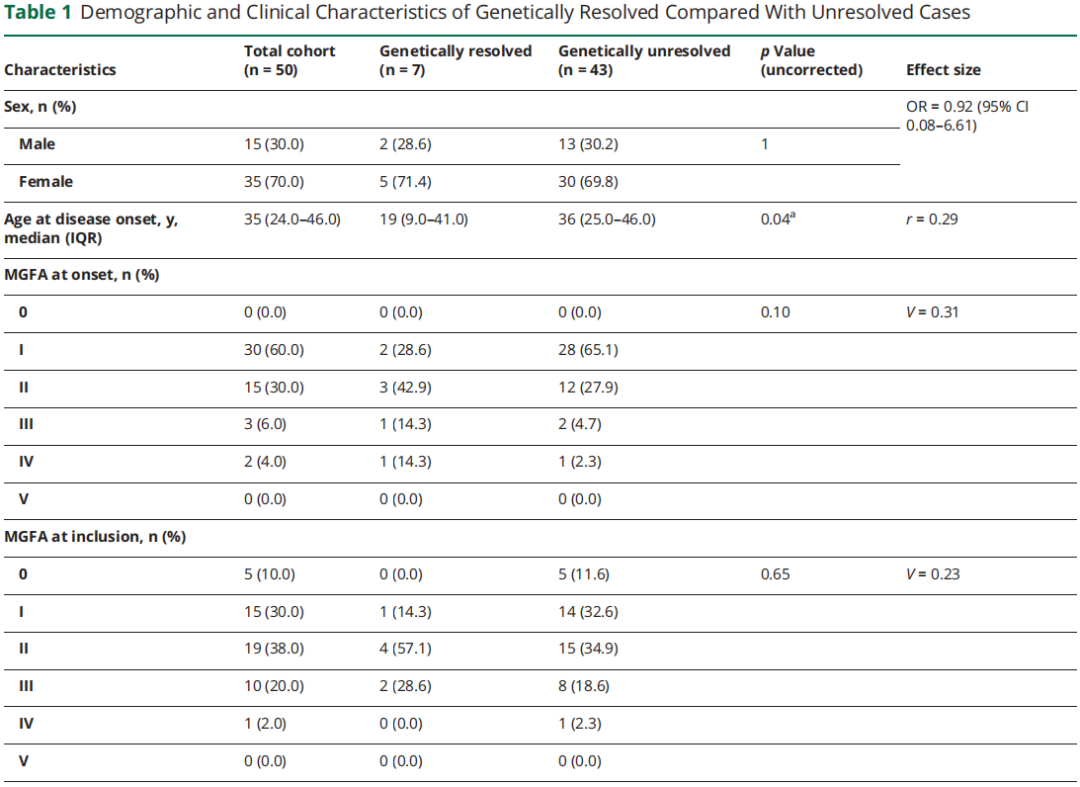
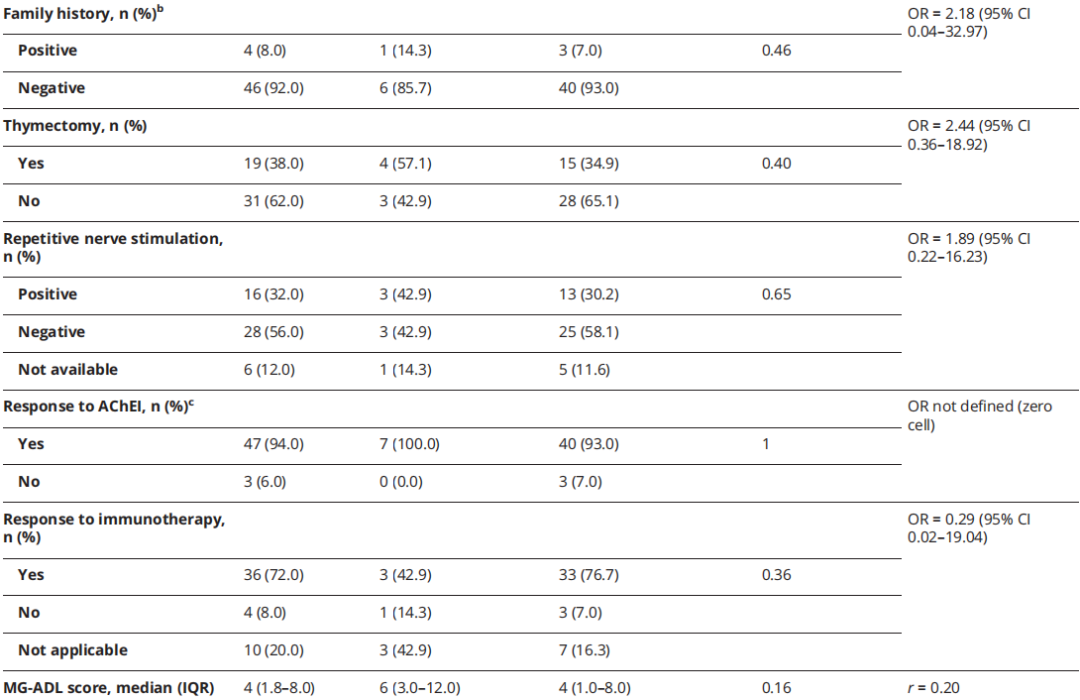

表1
基因诊断结果:
如图 2 所示,7 例患者检出 CMS 分子诊断,总诊断率 14%。3 家中心诊断率分别为 4/35(11.4%)、1/6(16.7%)、2/9(22.2%)。所有确诊病例均为常染色体隐性遗传,4 例复合杂合变异,3 例纯合变异。分子病因包括 CHRNE(n=4)与 RAPSN(n=3)。检出 7 种不同致病变异,包括 2 种错义变异、2 种剪接位点变异、2 种起始密码子丢失变异、1 种移码变异。根据 ClinVar 数据库,其中 5 种变异既往报道为(疑似)致病。2 种未报道变异依据以下 ACMG 标准判定为疑似致病:(1)CHRNE(NM_000080.4):c.2del(p.Met1?):PS1,PVS1_moderate,PM2_supporting;(2)RAPSN(NM_005055.5):c.169G>T(p.Gly57Cys):PM3_moderate,PP3_moderate,PS4_supporting,PM2_supporting。所有 CMS 病例的基因与临床特征列于表 2。
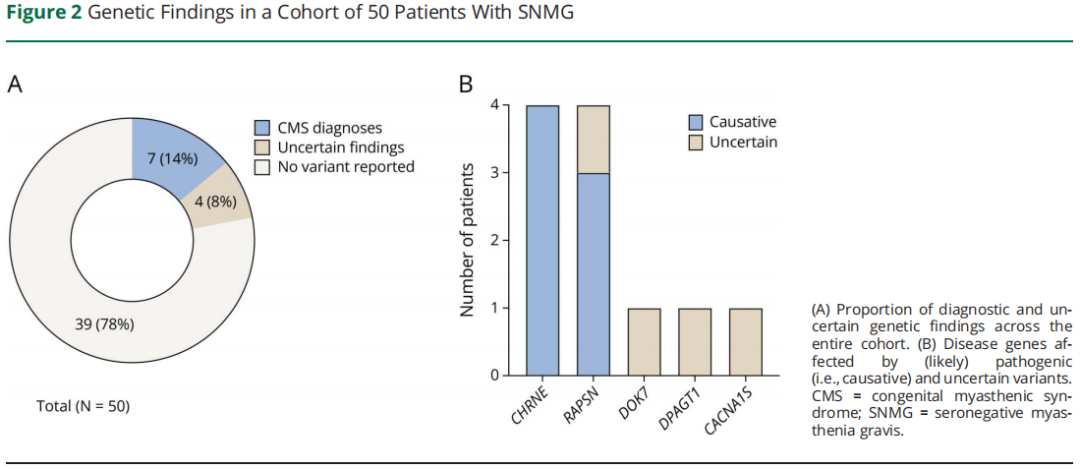
图2
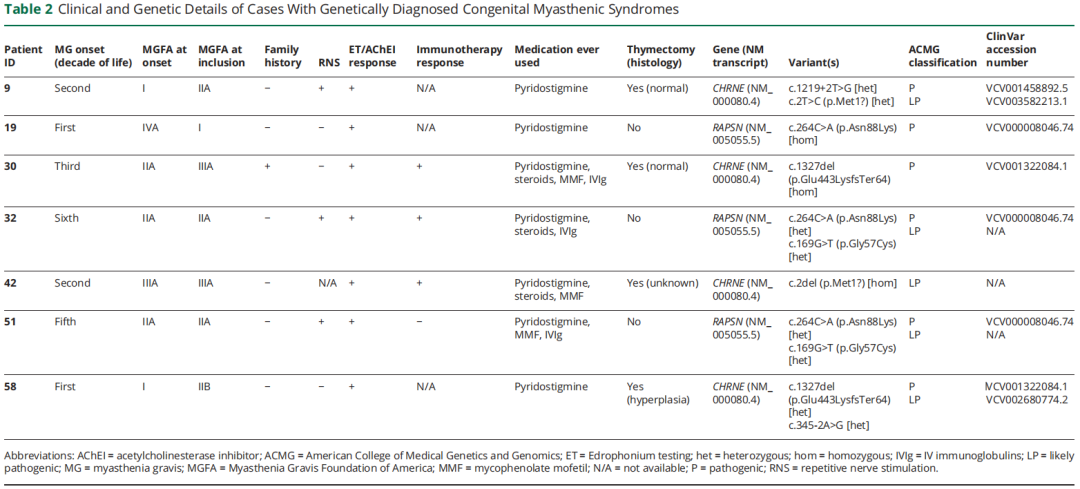
表2
分子诊断相关因素:
单因素分析显示疾病起病年龄与分子诊断相关(Mann-Whitney U 检验,z=-2.03,p=0.04,r=0.29),提示小至中等效应量。但经多重检验校正后,该关联无统计学意义。未发现其他临床或人口学参数与分子诊断显著相关(表 1)。值得注意的是,仅 1 例确诊病例有阳性家族史,但受累同胞的抗体状态或基因检测结果不详。重要的是,4 例分子诊断前接受免疫治疗的 CMS 患者中,3 例报告至少部分缓解。
临床意义未明的基因发现:
采用严格标准报告临床意义未明的发现(前述标准),本研究描述 4 例携带与肌无力表型关联不明的有趣变异。首先,1 例 30 多岁起病的眼肌型 SNMG 患者检出 RAPSN 杂合致病变异(NM_005055.5:c.264C>A,p.Asn88Lys)。该变异在普通人群中相对常见,基因组聚合数据库(gnomAD)等位基因频率约 0.2%,可能为本队列偶然检出。其次,1 例 20 多岁起病、全身型症状患者检出 DPAGT1 杂合截短变异(NM_001382.4:c.333_337del,p.Gly112CysfsTer3)。鉴于 RAPSN 与 DPAGT1 相关 CMS 为隐性遗传,这些变异的临床意义尚不明确。两例患者经 WGS 均未检出另一等位基因变异。此外,1 例以眼肌症状为主、30 多岁起病的 SNMG 患者检出 ClinVar 收录的 CACNA1S 杂合致病变异(NM_000069.3:c.3725G>A,p.Arg1242Lys)。CACNA1S 已知与 “1 型低钾型周期性麻痹”(OMIM #170400)、“5 型恶性高热易感”(OMIM #601887)相关,但与肌无力无关,该变异在 SNMG 中的临床意义尚不明确。最后,1 例全身型 SNMG 患者检出 DOK7 罕见纯合 VUS(NM_173660.5:c.971C>T,p.Pro324Leu),表现为轻度易疲劳,20 多岁早期起病。重复神经刺激示病理性递减,患者接受胸腺切除术(组织学:轻度胸腺增生)。最近随访时患者无症状且停药(MG-ADL 评分:0)。尽管 gnomAD 数据库无纯合携带者,但多种生物信息学工具预测该 DOK7 变异为良性(如 AlphaMissense 评分 = 0.099 [“疑似良性”],RareExome 变异集成学习器评分 = 0.29)。临床中该患者从未出现 DOK7-CMS 典型的肢带肌无力或行走困难。综上,该变异与 MG 表型的因果关系可能性较低。
讨 论
既往研究初步提示,部分 SNMG 病例由 CMS 相关基因单基因变异所致。鉴于自身免疫性 MG 与 CMS 治疗方案不同,病因区分在临床中至关重要。为解决该问题,本研究对 50 例特征明确的 SNMG 患者队列应用 WES,确诊 14% 的 CMS 分子诊断。检出 CMS 相关基因 CHRNE 与 RAPSN 致病变异,其表型与自身免疫性肌无力综合征难以区分。但本队列 CMS 病例的基因谱仅局限于 2 个基因,这一结果出乎意料。根据既往证据,其他类型 CMS(如 DOK7 相关、糖基化缺陷、慢通道综合征)也可能被误诊为 SNMG,更大队列研究有助于更全面揭示未确诊 CMS 病例的遗传背景。
值得注意的是,最终经 WES 确诊 CMS 的患者中,多数(86%)曾接受免疫治疗或胸腺切除术,这些治疗对 CMS 通常无效,还可能伴随严重不良反应与并发症。尽管如此,有报道部分最终确诊 CMS 的病例对免疫治疗有反应。鉴于 MG 临床试验中安慰剂反应率较高,研究者认为安慰剂效应是该现象最合理的解释。尽管如此,个别患者仍不能完全排除双重诊断(即自身免疫性 SNMG 合并 CMS)的极小可能。
本研究未发现分子诊断的统计学显著预测因素。单因素分析显示基因确诊病例起病年龄更小,但经多重检验校正后该关联无统计学意义。值得注意的是,本研究也观察到晚发型、症状轻微的 CMS 病例。7 例确诊病例中 5 例青少年或成年起病(1 例 60 多岁起病),仅 1 例 CMS 患者有阳性家族史。迄今为止仅有的一项 WES 研究局限于阳性家族史患者,而本研究未发现家族史是预测因素。但本研究样本量可能不足以稳健识别分子诊断的预测因素(如起病年龄小、阳性家族史)。
除明确致病变异外,本研究还报告 4 例临床意义未明的基因发现,包括 CACNA1S 致病变异 —— 该神经肌肉疾病基因既往与肌无力表型无关。重新表型分析未捕获到 CACNA1S 相关临床特征,因此该变异为偶然发现还是 CACNA1S 表型扩展尚不明确。另外 3 例意义未明发现包括隐性遗传 CMS 基因(RAPSN、DPAGT1)杂合致病变异与 DOK7 纯合 VUS,需进一步随访研究明确。
本研究的主要优势为前瞻性、无偏倚设计。患者经全面诊断流程预选,包括采用放射免疫法与细胞法全面筛查所有 MG 相关致病抗体,系统性对 SNMG 队列应用 WES。所有患者均由不同三级医疗中心经验丰富的神经肌肉专科医生临床评估,这些中心基因检测阈值通常较低。尽管如此,重要的是,所有患者既往均未接受常规临床基因检测。
本研究的局限性在于 SNMG 缺乏公认的诊断标准,SNMG 诊断建议近期才发布。值得注意的是,高灵敏度诊断工具单纤维肌电图(SFEMG)在奥地利未常规应用,而重复神经刺激与腾喜龙试验灵敏度有限。我们承认,奥地利队列的诊断率可能无法外推至诊断体系不同的国家(如常规应用 SFEMG 的国家)。由于缺乏高灵敏度诊断工具,部分 MG 模拟病例可能被纳入本研究。既往研究证实 MG(尤其是 SNMG)易误诊,功能性神经系统疾病是最常见的替代诊断。排除潜在模拟病例可能使基因诊断率更高。
尽管其他研究采用更严格的纳入标准(如重复神经刺激病理性递减或对 AChEI 有临床反应),但本队列仅 1 例患者不符合这些标准(该患者仅主观对免疫治疗有反应)。因此,应用这些标准不会显著影响本研究诊断结果。值得注意的是,将治疗反应作为纳入标准可能低估难治性患者比例,潜在导致选择偏倚。此外,药物临床反应评估常基于患者主观报告与评估医生判断,易受安慰剂效应影响。这凸显了该人群需要更客观的治疗反应评估指标。最后,尽管尝试联系中心数据库所有符合条件的患者,但部分患者无法联系或拒绝参与,可能引入额外偏倚。
另一局限性与 WES 固有的方法学缺陷相关,包括对重复扩增、结构变异与深度内含子变异的检测能力有限。尽管 CMS 基因 panel 可覆盖本研究所有诊断变异,但既往神经肌肉遗传病数据提示,WES 等无偏倚方法相比靶向检测具有显著优势,尤其对于非典型表型。随着成本持续下降,全基因组测序(WGS)与多组学技术等更全面的方法凭借技术优势,将进一步助力 WES 阴性病例的诊断明确。尽管尚无 CMS 不同基因检测应用的特异性成本效益数据,但儿科神经肌肉疾病研究显示,二代测序相比传统方法可降低成本。
参考文献:
Krenn M, Wagner M, Schuller H, Pugna I, Rath J, Zulehner G, Keritam O, Weng R, Koneczny I, Schiavo E, Damato V, Kleinveld VEA, Kiss C, Gold V, Quasthoff S, Masi G, O'Connor KC, Canning J, Waters PJ, Lenz D, Blüthner M, Pavlov M, Graf E, Winkelmann J, Löscher WN, Zimprich F, Cetin H. Screening for Congenital Myasthenic Syndromes in Adults With Seronegative Myasthenia Gravis Using Next-Generation Sequencing. Neurology. 2025 Oct 21;105(8):e214177. doi: 10.1212/WNL.0000000000214177. Epub 2025 Sep 26. PMID: 41004697.