深度解析医学证据,DeepEvidence为你支撑决策
肺癌是全球癌症相关死亡的首要原因。对于晚期EGFR突变非小细胞肺癌(NSCLC)患者,EGFR酪氨酸激酶抑制剂(EGFR-TKIs)是首选治疗方案,但获得性耐药难以避免,多在用药10—24个月后出现。因此,探索TKI耐药的分子机制并寻找新的治疗策略具有重要的临床意义。
近日,郑州大学的Bing-Hua Jiang/Lin Wang教授团队在本刊发表了题为“PDK1 elevation was induced by epigenetic modifications of KDM3A and METTL16 to mediate TKI resistance and cancer development”的研究论文。该研究揭示了KDM3A/METTL16/PDK1轴通过表观遗传修饰介导EGFR-TKI耐药的机制,并证实PDK1抑制剂JX06与吉非替尼联合治疗具有协同抗肿瘤效应。
研究团队首先发现,PDK1(丙酮酸脱氢酶激酶1)在吉非替尼耐药细胞系(PC-9/G、HCC827/OR)中表达显著上调。PDK1敲除使细胞对TKI治疗更敏感,而PDK1过表达则使敏感细胞产生耐药。功能实验表明,PDK1过表达促进葡萄糖消耗和乳酸产生,并增强细胞增殖能力。TCGA数据库分析显示,PDK1高表达与肺癌患者预后不良相关。
机制研究揭示了一条表观遗传调控轴:组蛋白去甲基化酶KDM3A通过H3K9去甲基化促进PDK1转录;同时,KDM3A上调RNA m6A甲基转移酶METTL16,后者通过m6A修饰增强PDK1 mRNA稳定性(图1)。研究还发现IGF2BP1作为m6A阅读蛋白识别并稳定修饰后的PDK1 mRNA。
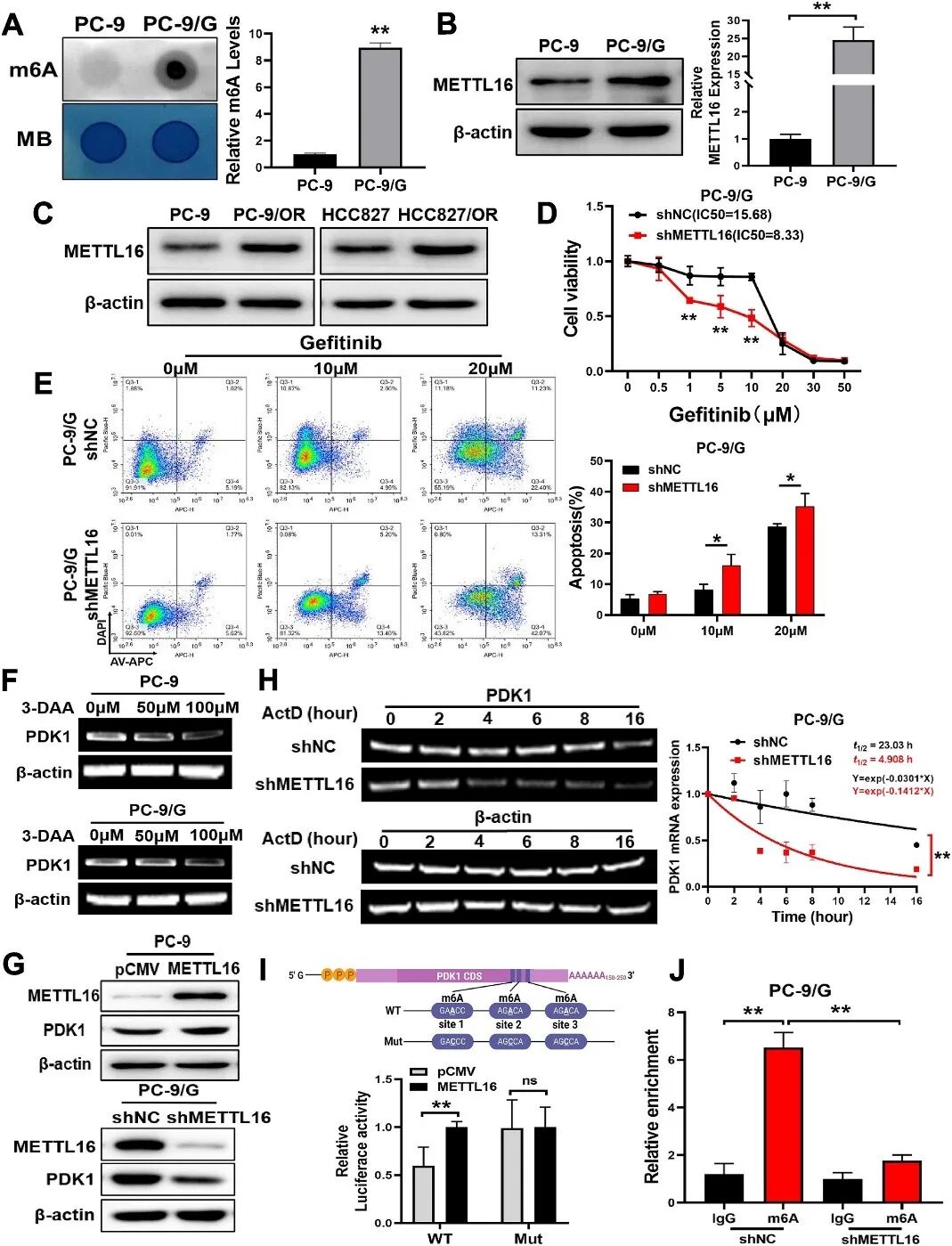
图1 METTL16通过m6A修饰增强PDK1 mRNA稳定性(原文中Figure 4)
在治疗策略探索中,研究团队发现PDK1抑制剂JX06与吉非替尼在体外可协同诱导细胞凋亡。为验证其体内疗效,研究者建立了肺癌异种移植瘤模型。结果显示,JX06与吉非替尼联合治疗显著抑制肿瘤生长,肿瘤体积和重量均明显减小,且对正常细胞毒性较低(图2)。免疫组化染色显示,联合治疗组Ki67和CD31表达明显降低,提示肿瘤增殖和血管生成受到抑制。
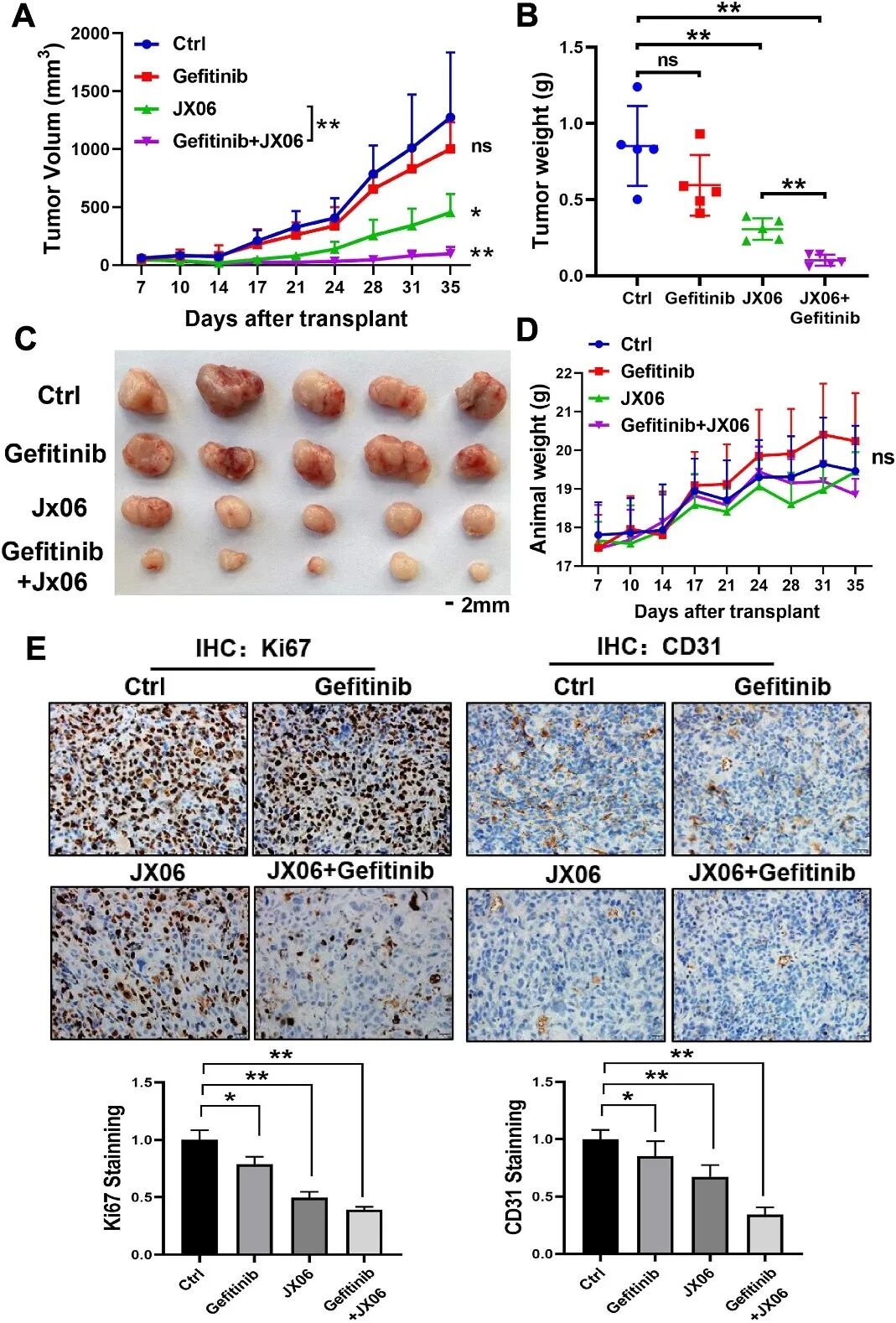
图2 JX06与吉非替尼联合治疗在体内显著抑制肿瘤生长(原文中Figure 7)
综上所述,该研究阐明了PDK1在EGFR-TKI获得性耐药中的关键作用及其表观遗传调控机制。KDM3A通过H3K9去甲基化促进PDK1转录,同时上调METTL16;METTL16通过m6A修饰增强PDK1 mRNA稳定性,由此形成表观遗传调控网络。PDK1抑制剂JX06与吉非替尼联合治疗具有协同抗肿瘤效应,为克服EGFR-TKI耐药提供了实验依据。
文章来源
免费全文下载链接:
https://www.sciencedirect.com/science/article/pii/S2352304225004362
引用这篇文章:
Zhou Z, Zhang R, Zhang Z, et al. PDK1 elevation was induced by epigenetic modifications of KDM3A and METTL16 to mediate TKI resistance and cancer development. Genes Dis. 2026;13(4):101947.