深度解析医学证据,DeepEvidence为你支撑决策
红细胞生成性原卟啉病(EPP)是一种罕见的常染色体隐性遗传病,由FECH基因突变导致亚铁螯合酶缺乏,进而使原卟啉在多个器官中蓄积引起。存在肝脏损伤的EPP患者的组织病理学表现不典型,这给病理诊断带来了挑战。本文报告一例 29 岁男性患者,该患者有反复发作的腹痛和巩膜黄染症状。初期肝功能异常提示为自身免疫性肝炎,但肝活检显示存在密集的棕褐色颗粒状沉积物,在偏振光下呈红色双折射,这是EPP的典型特征。基因检测发现了FECH基因一个新的突变位点(c.804+1del),该位点可能与本病相关。此外,该患者还患有吉尔伯特综合征。本病例凸显了EPP的诊断难点,以及结合临床病史、组织病理学检查和基因检测的重要性。偏振光显微镜检查对于识别EPP的特征性表现至关重要。早期基因检测可助力及时诊断和治疗,也有助于加深对这种罕见疾病的认识。
背 景
卟啉病是一类由血红素生物合成通路相关酶缺陷导致的遗传性代谢疾病。红细胞生成性原卟啉病(EPP)是一种常染色体隐性遗传病,因亚铁螯合酶活性丧失,破坏了血红素的生物合成,导致原卟啉在皮肤、血液、肝脏等多种组织中蓄积。这种蓄积引发了该病的特征性临床表现。EPP属于罕见病,国际研究报道其发病率为每百万人中 2 至 5 例。临床上,EPP主要表现为皮肤敏感性增高,少部分患者会出现肝脏受累。EPP患者的肝脏大体检查通常呈深色外观,镜下分析可见胆汁淤积改变以及双折射沉积物,该沉积物在偏振光显微镜下会形成特征性的马耳他十字结构。本报告描述了一名因肝功能异常入院的EPP患者病例,经详细病史核查、肝活检及全外显子组测序后确诊。此外,在FECH基因中发现了一个新的突变位点c.804+1del,该突变可能与本病的发生相关。
病 例
患者男,29 岁,因反复发作的腹痛及巩膜黄染,先后于 2022 年 4 月、2023 年 12 月、2024 年 9 月多次入院。首次入院时即发现肝功能异常,实验室检查结果如下:丙氨酸氨基转移酶(ALT)412 U/L(参考范围:0–40 U/L),天门冬氨酸氨基转移酶(AST)201 U/L(参考范围:0–40 U/L),总胆红素 221.1 μmol/L(参考范围:0–6.8 μmol/L),直接胆红素 118 μmol/L(参考范围:3.4–17.1 μmol/L),白蛋白 41.2 g/L(参考范围:35–55 g/L)。自身免疫性肝炎抗体检测结果如下:抗核抗体(ANA)(1:40)阳性(核仁型),抗核抗体(ANA)(1:80)弱阳性(核仁型),抗核抗体(ANA)(1:100)弱阳性(核仁型),平滑肌抗体阴性,线粒体抗体阴性。患者既往史无特殊,自述存在“光敏感”家族史,但家族中未发现类似疾病病例。入院时患者皮肤及巩膜重度黄染,腹部柔软,伴局部压痛,无反跳痛。常规腹部超声显示胆囊底部存在低回声区,提示胆汁淤积及胆泥形成。腹部CT血管造影(CTA)显示门静脉高压、食管下段静脉曲张、脾大、肝实质密度不均,以及肝内胆管轻度扩张(图1)。

▲图1 可见肝实质密度不均匀(红色箭头所示),伴有脾肿大,脾脏外缘延伸超过五个肋骨单位(蓝色箭头所示)
入院时,患者初步诊断为不明原因肝功能异常。随后开展了系统检查以排除肝损伤的常见病因:患者否认饮酒史;乙型肝炎病毒血清学标志物、人类免疫缺陷病毒(HIV)抗体、丙型肝炎病毒抗体、甲型肝炎病毒抗体、戊型肝炎病毒抗体均为阴性,排除病毒性肝炎可能;甲胎蛋白(AFP)检测未见异常,排除肝癌;自身抗体检测(包括抗肾小球基底膜抗体GBM、抗蛋白酶3抗体PR3、抗髓过氧化物酶抗体MPO等)排除了系统性疾病或结缔组织病相关的肝损伤。由于自身免疫性肝炎相关自身抗体呈部分阳性,暂无法排除自身免疫性肝炎。基于上述情况,对患者行肝活检以进行病理检查。活检获取到长约 7 mm、直径约 1 mm的黑色肝组织样本。镜下可见肝小叶结构破坏,汇管区纤维组织增生,导致肝细胞簇形成;间质内可见炎症细胞浸润,伴随胆管增生,肝细胞呈轻度水样变性及轻度脂肪变性(图2a)。肝窦及肝细胞内可见密集的棕褐色颗粒状沉积物(图2b)。在偏振光显微镜下,该颗粒沉积物显示出红色双折射,呈现特征性的马耳他十字结构(图2c)。免疫组化染色结果显示,CD138、CD4、CD8、IgG、IgG4及IgM均为阴性,小胆管CK7、CK19呈阳性。特殊染色结果显示,Masson染色阳性,铁染色、铜染色、糖原染色均为阴性(图3a–d)。最终病理诊断为红细胞生成性原卟啉病(EPP)合并肝硬化。
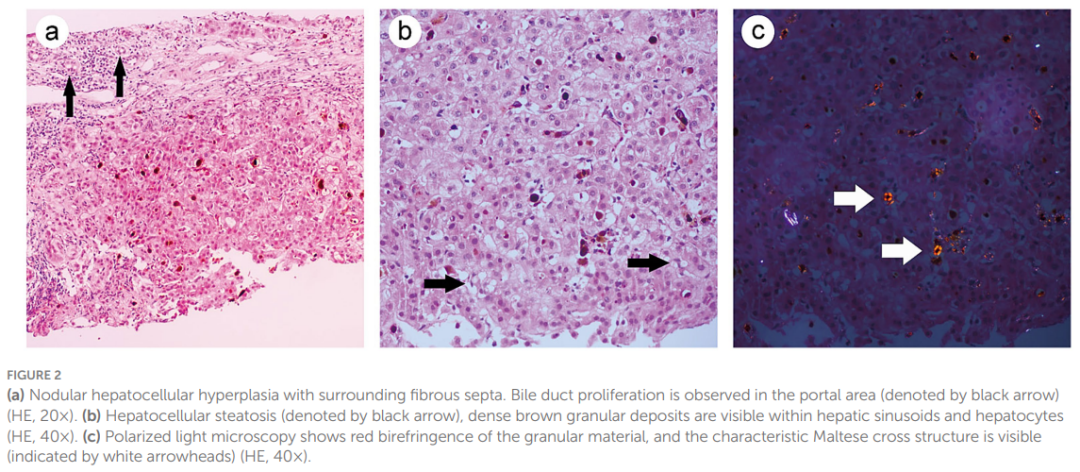
▲图2(a)结节状肝细胞增生伴周围纤维间隔。门静脉区可见胆管增生(黑箭头所示)(HE染色,20倍);(b)肝细胞脂肪变性(黑箭头所示),肝窦和肝细胞内可见致密的棕色颗粒状沉积物(HE染色,40倍);(c)偏振光显微镜下可见颗粒状物质呈红色双折射,并可见特征性的马耳他十字结构(白箭头所示)(HE染色,40倍)
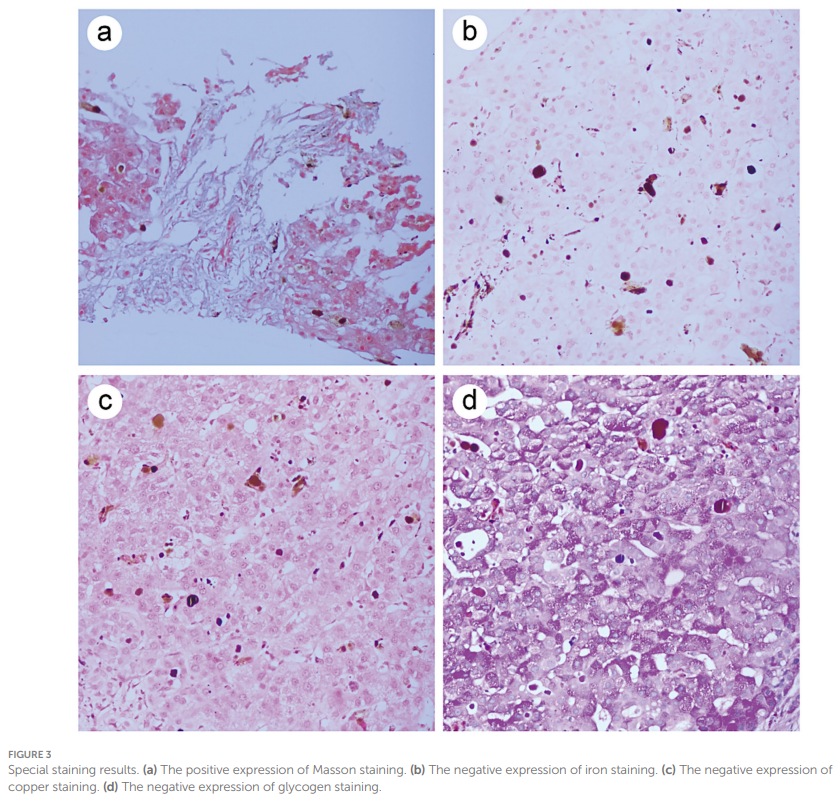
▲图3 特殊染色结果
随后,建议患者行卟啉病相关基因检测。本次基因检测采用基于靶向序列捕获的高通量测序技术开展全外显子组测序(WES),参考基因组为人类参考基因组组装版本GRCh38(hg38)。检测结果显示,患者FECH基因存在 2 个杂合变异:c.804+1del和c.315-48 T>C。此外,还检测到患者UGT1A1基因上游调控区存在杂合单核苷酸替换c.-3275 T>G,编码区存在杂合错义突变c.211G>A:p. G71R(图4)。最终基因诊断为:FECH基因异常导致常染色体隐性遗传EPP,同时合并UGT1A1基因异常导致常染色体隐性遗传吉尔伯特综合征。
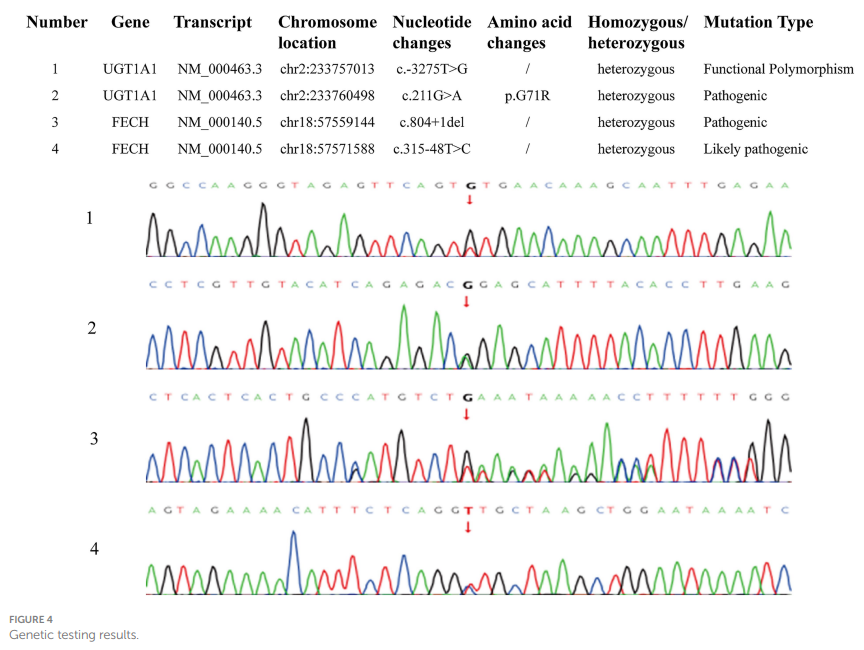
▲图4 基因检测结果
临床建议患者做好光防护,同时开始口服甲泼尼龙片(24 mg,每日 1 次)、沙利度胺(25 mg,每日 3 次),并辅以多烯磷脂酰胆碱保肝治疗。经过一段时间的支持治疗,患者病情稳定,黄疸消退。出院时,嘱患者严格做好光防护,若出现光敏感表现,可考虑口服β-胡萝卜素补充治疗;同时强调需避免饮酒。安排患者定期监测肝功能及乙型肝炎相关血清学标志物,出院后持续随访。
讨 论
EPP是一种常染色体隐性遗传病,由血红素生物合成通路中的亚铁螯合酶活性缺乏所致,会导致原卟啉在皮肤、血液、肝脏等组织中蓄积,进而引发相关临床症状。EPP的临床表现以皮肤敏感性增高为主要特征,少数患者会出现肝脏损伤。EPP患者的肝脏大体观察呈深色,镜下可见胆小管、肝细胞及库普弗细胞内存在密集的棕红色原卟啉聚合物结晶。这些结晶在偏振光显微镜下具有双折射性,呈现“星空样”外观,部分可显示特征性的马耳他十字结构。此外还可见不同程度的肝细胞变性坏死,病情更严重者可出现不同程度的纤维化及肝硬化。
本例患者多次入院,表现为腹痛及巩膜黄染。实验室检查提示肝功能异常,且自身免疫性肝炎抗体部分呈阳性。临床最初考虑为自身免疫性肝炎,送检肝活检至病理科。苏木精-伊红(HE)染色显示肝小叶结构破坏,汇管区纤维组织增生,小叶分隔导致肝细胞簇形成,肝细胞及肝窦内均可见胆汁淤积改变。病理科医师反复仔细阅片后,行免疫组化及特殊染色检查,结果显示Masson三色染色阳性,小胆管表达CK7和CK19,符合肝硬化表现;CD138、CD4、CD8、IgG、IgG4及IgM均为阴性,排除自身免疫性肝炎;特殊染色结果显示铁、铜、糖原染色均为阴性,进一步排除血色病、威尔逊病及糖原贮积症。此时患者的病因仍不明确。镜检发现肝细胞及肝窦内的胆栓存在形态学异常:这些类似胆栓的棕褐色物质颜色更深、结构更致密,而典型胆栓表现为均质、浅染,无颗粒样物质。组织活检报告提及肝标本存在异常的黑色外观,结合患者年龄相对较轻,病理医师怀疑为卟啉病。经反复询问病史,患者告知临床团队其存在“皮肤光敏感”家族史,进一步提示卟啉病可能。病理医师随后在偏振光显微镜下观察这些棕褐色颗粒,可见其呈现红色双折射,部分颗粒显示特征性的马耳他十字结构,符合卟啉病的病理特征。
鉴于卟啉病分类复杂,建议行基因检测。结果显示FECH基因存在异常,包含 2 个杂合变异:c.804+1del和c.315-48 T>C。EPP主要由FECH基因突变导致,本例患者为常染色体隐性遗传,上述突变所致疾病的临床特征与患者的“皮肤光敏感”家族史、腹痛、巩膜黄染等症状相符。值得注意的是,全外显子组测序还发现FECH基因存在一个经典剪接位点突变(NM_000140.5: c.804+1G>C),该变异为FECH基因804号内含子+1位的鸟嘌呤(G)被胞嘧啶(C)替换,该位点是供体剪接位点的高度保守核心核苷酸,因此该变异预计会破坏正常RNA剪接,通过异常转录本加工导致功能丧失(LOF)。该变异尚未在gnomAD人群数据库中被报道,根据美国医学遗传学与基因组学学会(ACMG)和临床基因组资源(ClinGen)制定的指南,该变异被分类为致病性变异,预计会对FECH蛋白的结构和功能产生不良影响。此外,还发现患者存在UGT1A1基因突变,包括上游调控区的杂合单核苷酸替换变异(c.-3275 T>G)和编码区的杂合错义突变(c.211G>A:p. G71R)。UGT1A1基因突变会导致常染色体隐性遗传的吉尔伯特综合征。吉尔伯特综合征又称先天性非溶血性黄疸或遗传性非结合胆红素血症,是一种胆红素代谢障碍疾病,通常起病隐匿,多无明显症状,临床特征为非结合胆红素升高。该病不会损伤肝细胞或胆管细胞,因此肝酶(ALT、AST)和胆道酶(ALP、γ-谷氨酰转移酶)通常正常。本例患者直接胆红素(118 μmol/L)和总胆红素(221.1 μmol/L)均升高,提示间接胆红素升高,符合吉尔伯特综合征的临床特征。但患者同时存在肝酶升高(ALT 412 U/L,AST 201 U/L),不能排除为EPP合并肝硬化所致。由于经济原因,患者未进行家系遗传谱系检测。
综上,本例患者临床表现不典型,无明显皮肤损害;受检测条件限制,未行血液、尿液及粪便原卟啉的定量检测,显著增加了诊断难度。最终通过病理医师与临床团队的密切协作,怀疑为EPP,并经基因检测确诊。EPP是一种罕见的遗传代谢病,容易出现误诊,要求病理医师具备丰富的诊断经验。在常规病理诊断中,当观察到肝窦或肝细胞内存在密集的棕色颗粒时,除考虑胆栓形成外,还应考虑其他代谢性肝病,如威尔逊病、糖原贮积症、血色病等,可通过特殊染色进行鉴别。EPP的诊断需要病理医师结合患者病史、临床表现及实验室检查结果进行综合判断。当怀疑EPP时,应采用偏振光显微镜观察是否存在特征性病理表现,同时尽早行基因检测,确诊后及时开展积极治疗,以改善患者预后。
参考文献:
Yang T, Chen C, Li C and Wang J (2025) Challenges in the pathological diagnosis of erythropoietic protoporphyria: a case report. Front. Med. 12:1664961. doi: 10.3389/fmed.2025.1664961