首页 > 健康资讯/ 正文
深度解析医学证据,lxfs.net为你支撑决策
论坛导读:肌张力障碍是一种以持续性或间歇性异常运动、姿势或二者兼有为特征的运动障碍性疾病,其临床诊断常因表现多样、病因复杂而延迟。近年来,随着诊断标准的更新、分类体系的完善以及对解剖网络、生理机制和分子通路认识的深入,肌张力障碍的研究取得了显著进展。2026年6月1日最新发表于《Lancet Neurol》的一项综述系统梳理肌张力障碍的临床表现、鉴别诊断、分类体系、病理生理机制(特别是神经发育起源)及治疗策略,强调尽管不同亚型之间存在显著的临床异质性,但共享的解剖网络、生理异常和分子通路为开发广谱治疗策略提供了可能,同时基因特异性变异也为个体化治疗开辟了新方向。

肌张力障碍(dystonia)是运动障碍门诊中常见的疾病类型,但误诊和延迟诊断现象普遍存在。其根本原因在于:临床表现高度异质——可为局灶性、节段性、多灶性或全身性,起病年龄从婴儿期到老年期不等,且常伴有震颤、肌阵挛等重叠运动现象;缺乏广泛适用的诊断性生物标志物,诊断主要依赖临床特征;遗传检测及其他辅助检查的可及性在全球范围内差异显著,限制了病因学诊断的明确。过去十年,通过细化临床表型特征、更新分类标准以及阐明潜在的解剖与生理机制,临床医生对肌张力障碍的识别能力已显著提升。
肌张力障碍的核心运动特征包括:异常姿势常呈模式化、重复性,可呈震颤样或抽搐样;症状通常由随意动作诱发或加重,并出现肌肉收缩向邻近或远端肢体“溢出”的现象;存在缓解性动作或感觉诡计,即患者通过特定随意动作或轻微触碰某一身体部位可暂时缓解肌张力障碍性收缩;放松或睡眠时症状减轻,醒后短暂持续;部分亚型(尤其是早发性多灶性或全身性肌张力障碍)对酒精反应良好。非运动表现同样是临床谱系的重要组成部分,包括疼痛、焦虑、抑郁、睡眠障碍、疲劳、轻度认知困难以及社交认知受损,这些非运动症状,特别是焦虑和抑郁,是患者生活质量的独立决定因素。目前尚不清楚非运动表现是原发症状还是继发于运动问题,但初步数据支持其具有原发性成分,因此临床评估应常规纳入对非运动症状的系统评价。
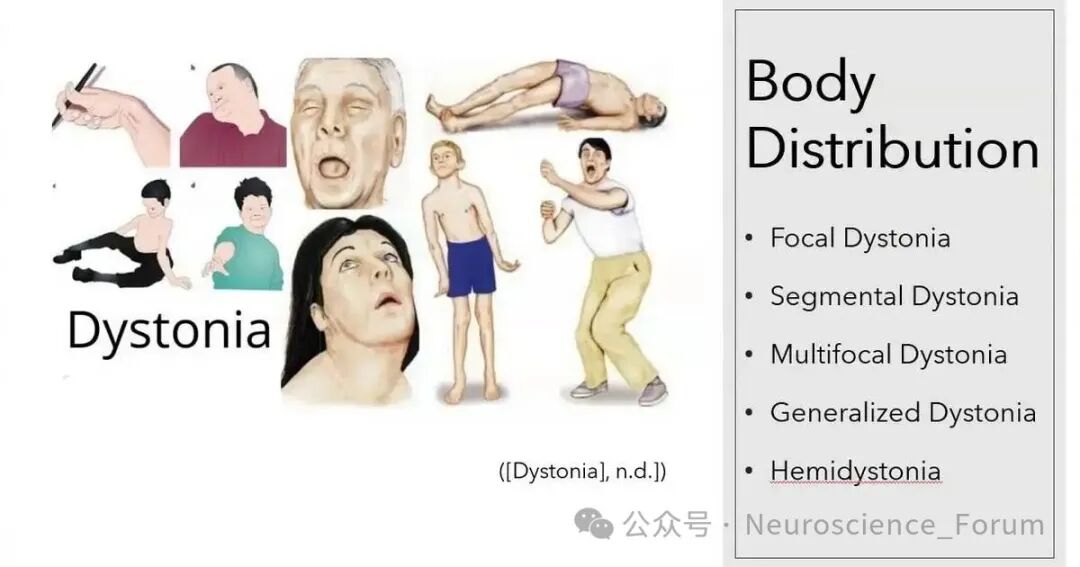
鉴别诊断方面,多种疾病可模仿肌张力障碍,包括功能性神经障碍、良性婴儿斜颈、Grisel综合征、僵人综合征及抗IgLON5病等。临床警示信号有助于区分,例如功能性障碍常表现为不恒定、可分散注意力的特征,而Grisel综合征多见于儿童、急性起病伴疼痛性斜颈且继发于头颈部感染或炎症。某些典型警示信号(如突发起病或强直姿势)偶尔也见于特定遗传性肌张力障碍(如快速起病的肌张力障碍-帕金森综合征),而情绪诱发的阵发性运动可能提示遗传性阵发性疾病或功能性运动障碍,需结合整体临床情境综合判断。
表1:肌张力障碍的临床诊断要点与鉴别诊断警示信号
|
临床特征 |
典型表现(支持肌张力障碍) |
警示信号(提示其他诊断或需谨慎鉴别) |
|
异常运动/姿势 |
持续性或间歇性,模式化、重复性,可呈震颤样或抽搐样 |
完全固定不变的姿势(提示结构性病变或纤维化);突发起病且快速进展(需警惕快速起病的肌张力障碍-帕金森综合征或功能性障碍) |
|
诱发与缓解因素 |
随意动作诱发或加重,存在“溢出”现象;放松/睡眠时减轻;感觉诡计(轻触特定部位)可短暂缓解 |
症状完全不能被任何因素改变;注意力集中时症状反而加重,分散注意力时减轻(提示功能性神经障碍) |
|
酒精反应 |
部分早发性多灶性或全身性肌张力障碍对酒精反应良好 |
酒精诱发或显著恶化运动障碍(需警惕其他遗传性阵发性运动障碍) |
|
非运动表现 |
常伴焦虑、抑郁、疼痛、睡眠障碍、疲劳,影响生活质量 |
突出且早于运动症状的认知功能减退、幻觉、睡眠行为障碍(提示抗IgLON5病等神经退行性疾病) |
|
病程与演变 |
缓慢进展,可向邻近身体区域扩散(如眼睑痉挛→口下颌/颈部) |
急性或亚急性起病,伴有感染/炎症病史(儿童需警惕Grisel综合征);出生后短期内出现且长期稳定不变(良性婴儿斜颈) |
|
体格检查线索 |
通常无明显肌肉萎缩或纤维化(长期严重者可继发) |
胸锁乳突肌萎缩、缩短、纤维化(提示良性婴儿斜颈或陈旧性损伤);被动活动时存在明确抵抗且无松弛(僵人综合征或Grisel综合征) |
|
特定综合征警示 |
— |
疼痛性颈/躯干僵硬且无异常运动(僵人综合征或PERM);伴有Kayser-Fleischer环、肝功异常(Wilson病);药物相关慢性暴露史(迟发性肌张力障碍) |
注:PERM:进行性脑脊髓炎伴强直和肌阵挛。部分警示信号(如突发起病)在极少数遗传性肌张力障碍中也可见,需结合完整临床情境判断。
分类体系:双轴框架的临床应用
根据最新共识,肌张力障碍采用双轴分类系统。轴I关注现象学和综合征识别,包括发病年龄、家族史、身体分布、时间维度(起病方式、病程、日内变异性)以及伴随特征。轴II聚焦病因与机制,分为遗传性和获得性两大类。局灶性肌张力障碍是成人起病的最常见形式,仅累及单一身体区域(如眼睑、口下颌、颈部、喉部、上肢、躯干或下肢)。疾病过程中症状可向其他区域扩散,国际队列研究显示:眼睑痉挛患者中58%扩散至口下颌或颈部,手部肌张力障碍扩散率为17%,喉部为16%,而颈肌张力障碍扩散率最低(8%)。
颈肌张力障碍是最常见的局灶形式,最新分类强调精细区分头颈部位置改变的具体方向(旋转、屈曲、伸展、侧倾、移位),以优化肉毒毒素局部注射的靶肌选择。任务特异性肌张力障碍是另一重要亚型,仅在执行特定熟练活动时触发,典型代表为音乐家肌张力障碍和书写痉挛,其发生可能与高度重复、超负荷的精细运动训练导致感觉运动系统可塑性失代偿有关。
发病机制的多层次整合
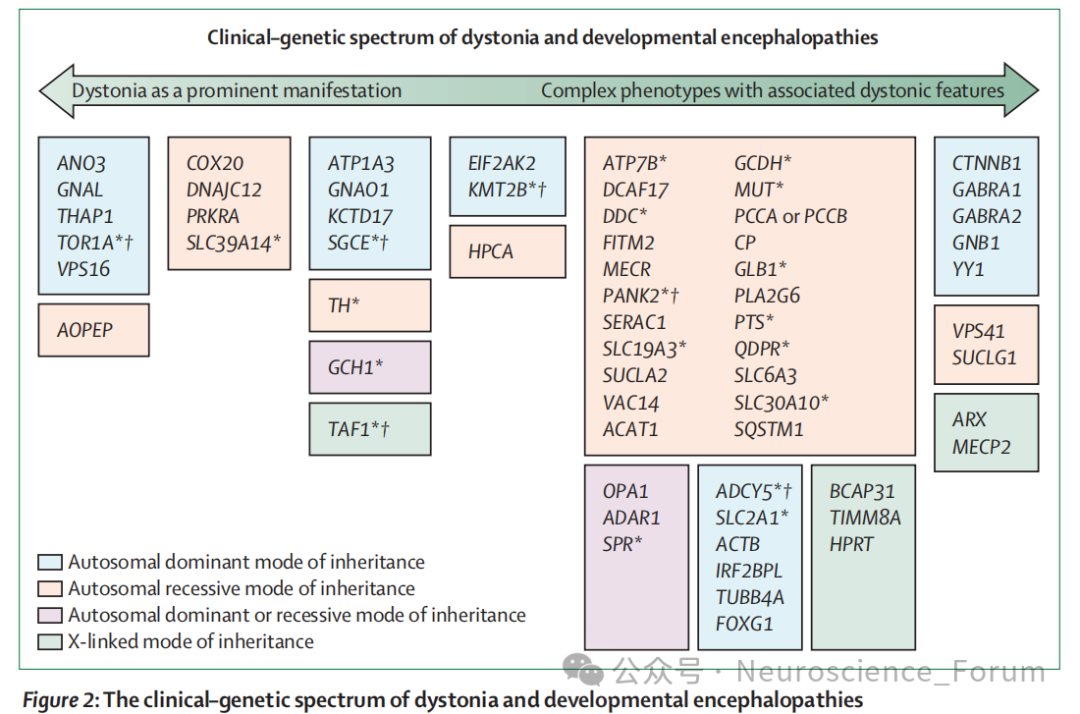
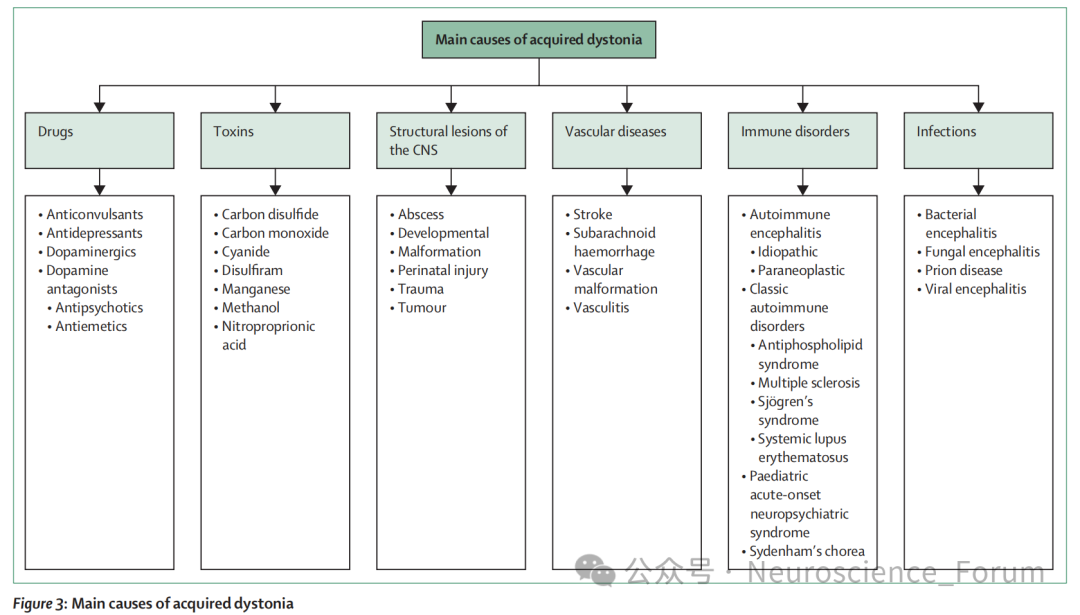
尽管肌张力障碍的病因高度异质——从数百种致病基因(图2)到获得性因素(药物、毒素、结构性病变、血管性疾病、免疫紊乱、感染,图3)——研究者仍致力于探寻共享的机制通路,这些机制可在分子、解剖、生理等多个层面发生汇聚。
解剖机制:肌张力障碍被认为是基底节、小脑、丘脑和感觉运动皮层构成的神经网络功能障碍所致。局灶性病变研究揭示了不同部位损伤与临床表型的关联:基底节局灶性病变最常导致肢体肌张力障碍,而小脑及脑干连接处病变更易引起颈肌张力障碍。即使在无明显结构性病变的患者中,正电子发射断层扫描也发现了重叠的脑区葡萄糖代谢异常模式,提示共享的解剖基础。
生理机制:主要包括中枢抑制与兴奋性神经递质失衡、感觉运动整合异常及适应不良性可塑性。在多种啮齿类肌张力障碍模型中,纹状体长时程抑制缺失伴随长时程增强增强是共同特征,多巴胺通路常被涉及(直接或间接)。小脑浦肯野细胞放电模式改变——从正常规律性简单棘波转为频繁簇状放电——在多个独立动物模型中被证实,进一步支持肌张力障碍是网络性疾病假说,即某一脑区功能障碍引起其他区域适应性改变,最终导致运动输出异常。
分子与细胞机制:多巴胺能神经传递异常是最深入研究且与临床最相关的通路。多巴反应性肌张力障碍通常由GCp基因变异导致多巴胺合成障碍,其他影响多巴胺合成、转运或突触后信号转导的基因(SPR、PRPS、TH、DDC、SLC6A3、SLC18A2、GNAL、GNAO1)也可引起肌张力障碍。非遗传机制如长期使用多巴胺受体拮抗剂导致的迟发性肌张力障碍、帕金森病左旋多巴诱导的异动症以及获得性脑损伤后的肌张力障碍均与多巴胺信号异常有关。然而,多巴胺并非唯一通路,其他已被识别的机制包括脑内金属(铁、铜、锰、钙)蓄积、细胞内运输障碍、嘌呤代谢异常、线粒体功能障碍及基因表达调控失常。
肌张力障碍作为神经发育障碍
近年来最具变革性的观点是将肌张力障碍视为神经发育障碍。在妊娠后期及生命最初几年,感觉运动皮层的组织结构与环路连接经历活动依赖性的精炼过程,此时期若发生感觉输入异常(无论源于遗传还是围产期损伤),可导致神经环路的永久性结构或功能缺陷,且成熟后的神经系统难以代偿。
动物实验显示,围产期缺氧-缺血及生后后肢活动限制可导致躯体感觉皮层地图退化及皮层兴奋性增高,这与局灶性手部肌张力障碍中观察到的皮层感受野重叠现象具有相似性。在人类,窒息新生儿的感觉诱发电位异常高度预测脑性瘫痪的发生,且部分肌张力障碍患儿即使头颅磁共振成像正常仍存在感觉诱发电位异常,提示感觉通路功能障碍可能是长期存在的并影响了早期感觉运动环路的发育建模。
TOR1A基因变异是最具特征的单基因肌张力障碍病因,其编码蛋白Torsin-1A在突触发生和突触可塑性中发挥重要作用。小鼠模型中,从胚胎期开始抑制Tor1A表达可诱导出DYT-TORIA表型,而成年后抑制则无此效应;同样,在幼年期(而非成年期)恢复Tor1A表达可挽救行为表型,有力证明了该基因的致病作用发生在关键神经发育窗口期内。DYT-TORIA患者中,年轻组(≤42岁)壳核乙酰胆碱转运体表达降低,而年长组则无此变化,为人脑中发育窗口期破坏提供了直接证据。
任何在关键发育时期剥夺脑网络正常活动的异常因素,不仅影响感觉运动环路的结构与连接,还会通过稳态可塑性机制改变其相对兴奋性。我们推测,在一些遗传性肌张力障碍中,发育窗口期的异常神经突发生被突触功能改变所代偿,进而引起可塑性反应及其范围的代偿性改变,从而强化了长时程增强样机制。对于围产期脑损伤导致的获得性肌张力障碍,感觉运动环路突然暴露于皮质-基底节-丘脑-皮质环或皮质-脑桥-小脑-丘脑-皮质环的深刻活动改变中,这种传入输入的相对(即使短暂)剥夺会改变这些环路功能的“设定点”,反映为另一种形式的适应不良性神经可塑性。在脑性瘫痪小鼠模型中,慢性(而非急性)纹状体胆碱能中间神经元兴奋可导致肌张力障碍表现,这与适应不良性可塑性假说一致。
大规模测序研究进一步揭示了肌张力障碍与发育性脑病之间的分子交集:经典发育性脑病基因的变异可表现为肌张力障碍表型;孤立性肌张力障碍相关基因的变异也可引起更广泛的发育性脑病特征;导致发育性脑病和肌张力障碍的基因变异可引发相似通路的 dysfunction。因此,孤立性肌张力障碍可被视为神经发育障碍表型连续谱的一端,而另一端则是无共病肌张力障碍的发育性脑病,中间存在大量混合表型。尽管这种重叠在儿童起病形式中最显著,但发育性脑病基因的变异在成人起病肌张力障碍中也被逐渐认识。临床上,癫痫-运动障碍综合征是这一连续谱的典型例证,约90%的患者伴有神经发育特征(主要为发育迟缓或智力残疾),其中肌张力障碍是最常见的运动障碍类型。
治疗靶点与策略
共享机制的揭示对实验性治疗具有重要意义,因为针对共享机制的治疗可能对多种肌张力障碍亚型有效。最典型的例子是肉毒毒素靶向过度的神经肌肉活动,无论病因如何均有效,但注射治疗限制了其在广泛受累患者中的应用。共享解剖机制为手术靶点提供了依据:苍白球脑深部电刺激对某些类型肌张力障碍高度有效,而丘脑靶点对其他类型更优,但仍有部分患者对两者均无反应,需进一步优化刺激位点。
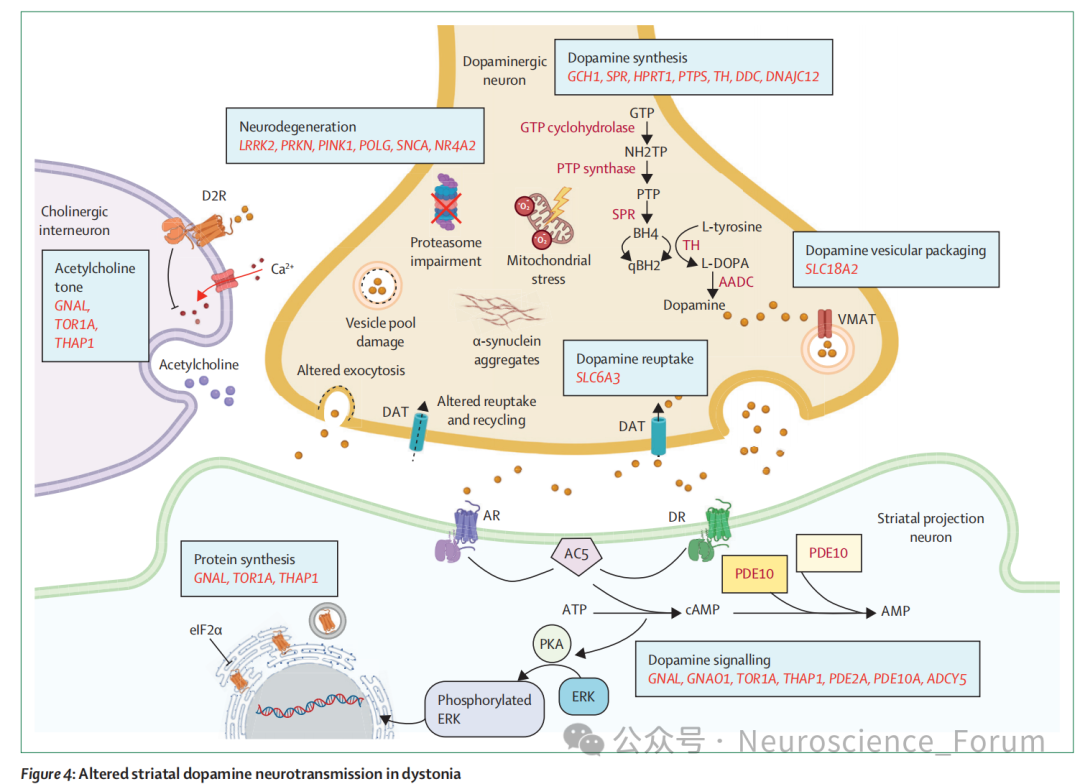
分子层面,多巴胺通路可通过多种方式调节,抗胆碱能药物和苯二氮䓬类药物在真实世界中对多种肌张力障碍具有广泛疗效,但需深入理解其机制以开发副作用更少、疗效更优的替代药物。更重要的是,理解共享机制在神经发育过程中的作用提示早期干预可能对某些亚型至关重要。同时,个体化治疗也日益受到重视:特定致病基因或变异可指导选择改变生命的治疗方案,如多巴反应性肌张力障碍的多巴胺治疗、GLUT1缺乏相关肌张力障碍的生酮饮食、ADCY5相关肌张力障碍的咖啡因、Wilson病的铜螯合或锌盐治疗,以及芳香族L-氨基酸脱羧酶缺乏症的基因治疗。
表2:肌张力障碍的共享发病机制及其对治疗的启示
|
机制层面 |
关键病理生理发现(原文证据) |
涉及的常见病因/基因或模型 |
潜在治疗靶点或策略 |
|
解剖网络 |
基底节、小脑、丘脑、感觉运动皮层构成的功能网络异常;基底节局灶病变→肢体肌张力障碍,小脑/脑干病变→颈肌张力障碍 |
获得性局灶性病变;特发性局灶性肌张力障碍(PET显示重叠的代谢异常) |
脑深部电刺激(苍白球/丘脑);网络靶向调控(如经颅磁刺激) |
|
生理机制 |
抑制-兴奋失衡;感觉运动整合异常;适应不良性可塑性(长时程抑制缺失、长时程增强增强);小脑浦肯野细胞簇状放电模式改变 |
TOR1A、THAP1、GNAL等多种遗传模型;音乐家/书写痉挛等任务特异性肌张力障碍 |
调节可塑性(如非侵入性神经调控);促进正常感觉运动整合(康复训练、感觉诡计训练) |
|
多巴胺能通路 |
突触前多巴胺合成/转运障碍(GCp、TH、DDC、SLC6A3等);突触后多巴胺受体信号转导异常(GNAL、GNAO1、ADCY5);多巴胺神经元变性(LRRK2、PRKN) |
多巴反应性肌张力障碍;Wilson病(间接影响);迟发性/药物性肌张力障碍;帕金森病相关异动症 |
左旋多巴(对多巴反应性高效);多巴胺受体拮抗剂(迟发性需谨慎);腺苷受体调节剂(如咖啡因对ADCY5相关有效) |
|
其他分子通路 |
脑内金属蓄积(铜、铁、锰、钙);细胞内运输障碍(核质转运、内质网、溶酶体);嘌呤代谢异常;线粒体功能障碍;基因转录失调 |
Wilson病(铜);泛酸激酶相关神经变性(铁);HPRT1(嘌呤);TOR1A(内质网/核转运) |
铜螯合剂/锌盐(Wilson病);铁螯合试验;生酮饮食(GLUT1缺乏);基因治疗(AADC缺乏) |
|
神经发育可塑性(共享视角) |
关键发育窗口期(围产期至幼年)感觉运动环路活动依赖性的建模/精炼异常;围产期缺氧/缺血、TOR1A基因胚胎期抑制等均可导致永久性环路缺陷;成熟后干预往往无效 |
脑性瘫痪相关肌张力障碍;DYT-TORIA;围产期脑损伤;部分早发性遗传性肌张力障碍 |
早期(幼年期)靶向干预(基因修复、康复、药物)恢复表型;避免成熟后不可逆改变;强调新生儿期风险因素监控 |
注:AADC:芳香族L-氨基酸脱羧酶;PET:正电子发射断层扫描。共享机制的识别有助于开发适用于多种肌张力障碍亚型的广谱治疗,同时基因特异性通路为个体化精准治疗提供依据。
研究空白与未来方向
尽管取得了显著进展,肌张力障碍领域仍存在诸多知识空白。临床诊断与异质性方面:早期识别仍具挑战,临床异质性的驱动因素知之甚少,缺乏普遍适用的生物标志物来确认诊断、区分亚型或预测疾病进展及治疗反应。神经生物学机制方面:基底节、小脑、脑干和皮层在发病中的相互作用尚未完全阐明,多巴胺能功能障碍的确切角色仍不明确,围产期脑损伤如何影响感觉运动环路发育及后期肌张力障碍风险需进一步研究,早期干预预防或改善神经元环路适应不良性发育的潜力尚未探索。遗传结构方面:许多基因变异的功能后果未完全理解,全球遗传图谱仍不完整,基因型-表型关联常不明确。
未来的研究重点应包括:通过大规模、多中心、表型充分鉴定的队列,结合多组学分析、多模态神经生理与神经影像,阐明基因型-表型关系;开发生物标志物以支持诊断、指导治疗和预测预后;系统表征非运动症状及患者报告结局;实施功能性研究以明确肌张力障碍相关遗传变异的作用;利用长读长测序等先进技术发现新的遗传原因(如结构变异和非编码变异);探究围产期脑损伤影响感觉运动环路成熟的机制及早期干预的关键窗口;阐明大规模网络改变如何导致症状和疾病进展;检查环境暴露和表观遗传机制对疾病起病和表型变异的作用。
结论
肌张力障碍的诊断仍主要依赖临床标准,多数形式缺乏可靠生物标志物。遗传学的重大进展显著提高了病因诊断能力,但基因变异的解读、外显率及检测可及性仍是未解决问题。肌张力障碍作为关键发育窗口期内感觉运动环路改变所致的神经发育障碍这一概念,以及众多潜在病因中存在共同机制的认识,为治疗开发(包括早期干预的可能性)指明了新靶点。个体化治疗策略的兴起也提出了医疗公平的重要问题——确保所有肌张力障碍患者都能获得适当的遗传检测及其他尖端诊断工具,避免错失治疗机会。未来的临床前和转化研究应同时靶向共享机制和基因/变异特异性效应,并加强对肌张力障碍网络神经生理活动变化的研究,以应对这一高度复杂疾病机制的挑战。
小提示:本篇资讯仅在梅斯医学APP中开放阅读,请扫描二维码直接下载APP
