深度解析医学证据,DeepEvidence为你支撑决策
tau蛋白病是以tau蛋白异常过度磷酸化并聚集形成包涵体为核心特征的神经退行性疾病,阿尔茨海默病是其中最具代表性的类型,tau聚集产生的神经毒性是驱动神经元死亡、认知衰退的关键因素。现有研究已发现神经元死亡参与tau相关神经退行性病变,但tau聚集引发神经元死亡的具体通路尚不明确,同时,转座因子、Z 型核酸、ZBP1 等分子在神经退行性疾病中的作用仍有待探索,tau病理、表观遗传异染色质稳态、内源核酸激活与神经元程序性死亡之间的调控关联也缺乏系统性阐释。
浙江大学莫玮教授、杨章华研究员、Qiang Chen、厦门大学韩家淮院士、张荧荧副教授等人以PS19 tau转基因小鼠为模型,系统探究tau聚集诱导神经元死亡的分子机制,明确tau神经毒性的关键介导通路,并挖掘可用于tau蛋白病干预的潜在治疗靶点。相关内容以“Tau aggregates cause reactivation of transposable DNA elements, leading to Z-RNA–ZBP1-mediated neuronal death”为题发表在《Nature Neuroscience》上。

【主要内容】
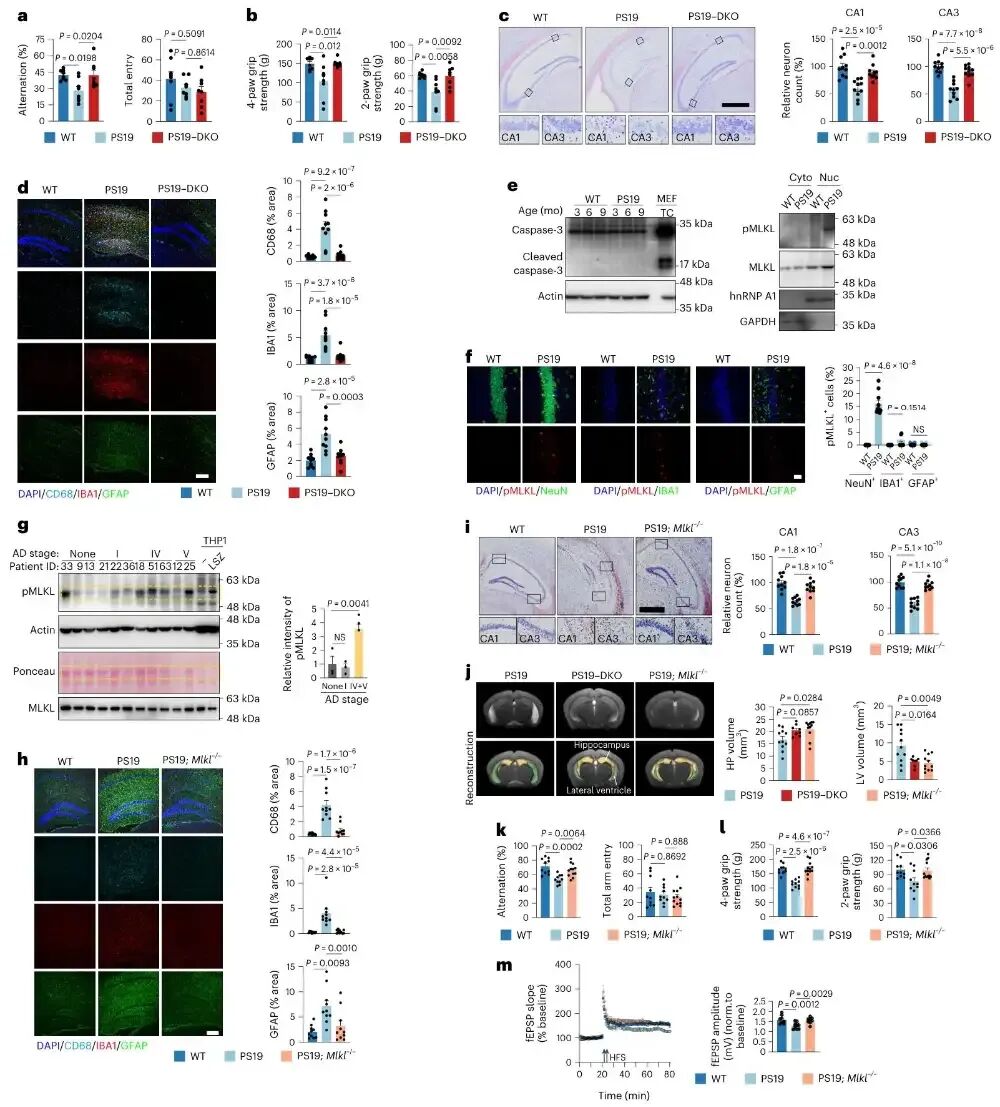
图1 神经元程序性坏死是tau介导神经变性的致病关键
作者通过对野生型小鼠、PS19 tau转基因小鼠以及Casp8/Ripk3双敲除、Mlkl敲除的PS19小鼠进行行为学、组织染色、蛋白检测和MRI成像分析,证实tau病理会特异性诱发神经元发生程序性坏死而非凋亡,结果显示阻断程序性坏死可显著改善PS19小鼠的认知与运动功能、减少神经元丢失、缓解神经炎症、恢复海马体积并逆转脑萎缩,同时在阿尔茨海海默病患者脑组织中也检测到程序性坏死标志物随疾病进展升高。
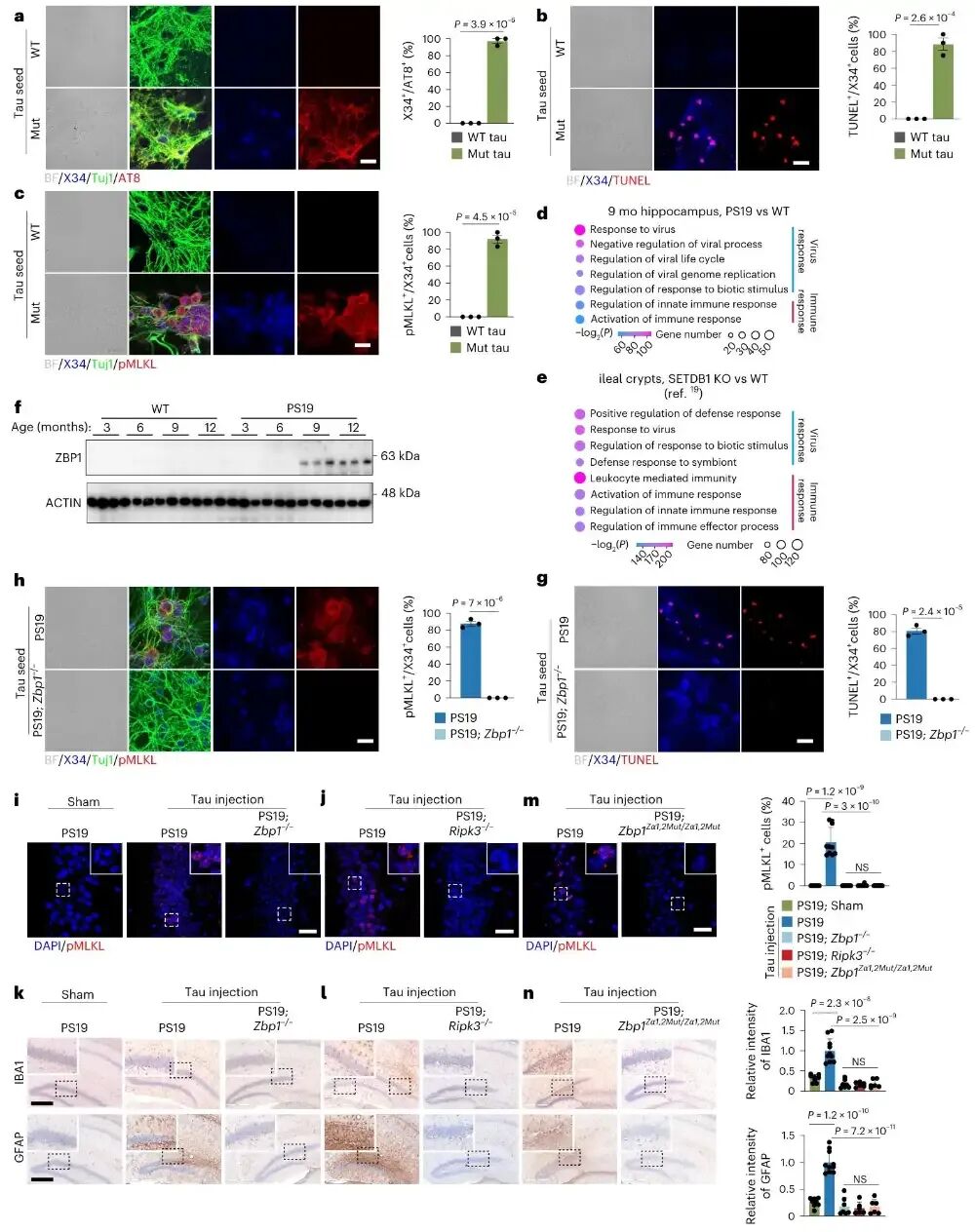
图2 tau聚集体通过内源性配体激活ZBP1诱发程序性坏死
作者在细胞与小鼠tau加速模型中证明,tau聚集体并非直接激活ZBP1,而是促使神经元产生内源性核酸物质作为“死亡配体”,转录组分析显示PS19小鼠的基因表达谱与ZBP1依赖的坏死通路高度相似,tau处理会使神经元出现ZBP1显著上调且主要定位在神经元中,敲除Zbp1或破坏其Zα结构域均可完全阻断tau诱导的神经元坏死与神经炎症。
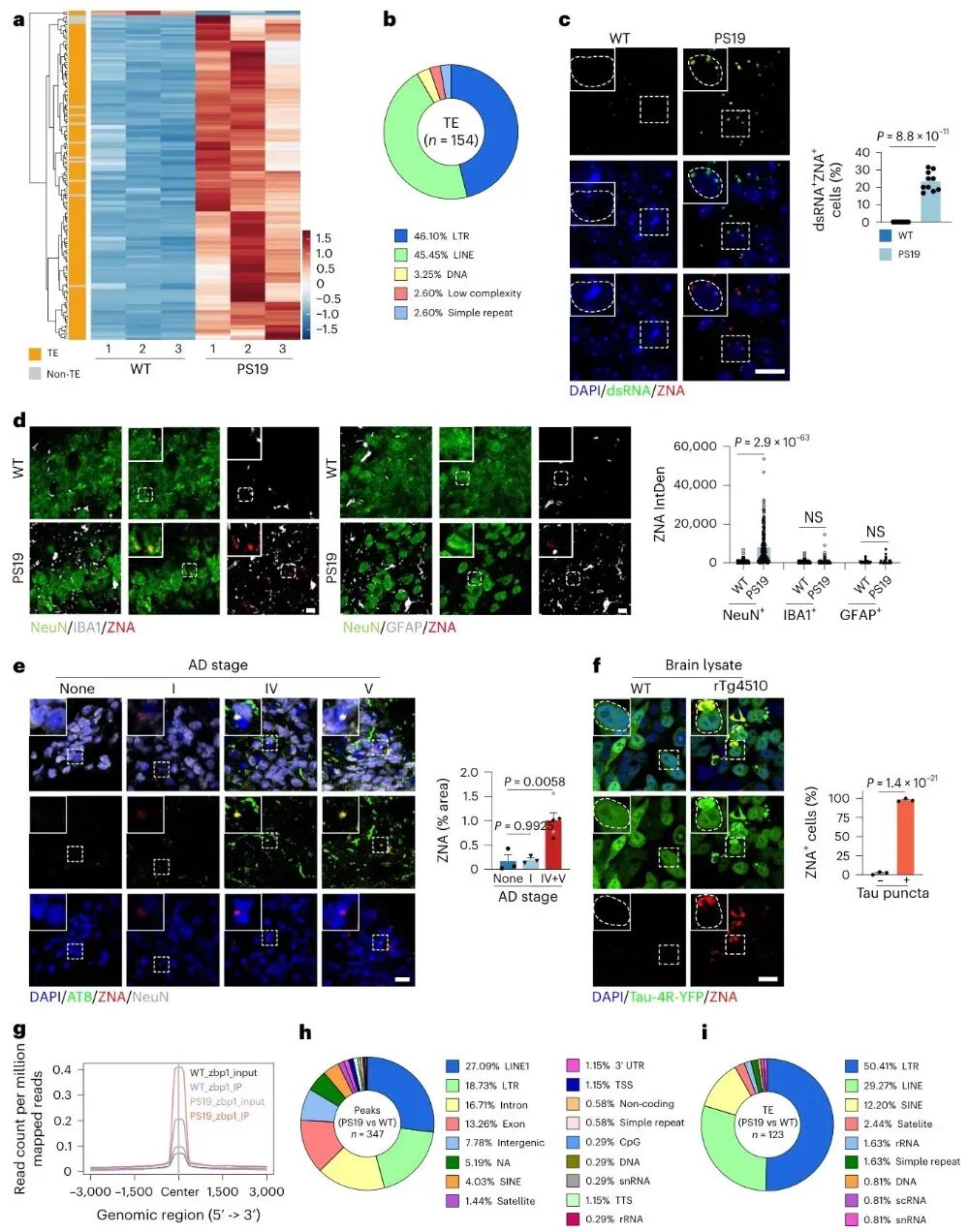
图3 致病性tau激活转座子产生Z-RNA并结合ZBP1
作者揭示tau聚集会导致神经元内转座因子(TE)异常激活,主要是LTR与LINE类型元件大量表达,并产生具有左手螺旋构象的Z型核酸(Z-RNA),Z-RNA信号主要富集在神经元中并与tau聚集体高度共定位,在阿尔茨海默病患者脑组织中同样检测到Z-RNA随疾病进展升高,RIP-seq进一步证实ZBP1能够特异性结合这些来自转座子的Z-RNA。
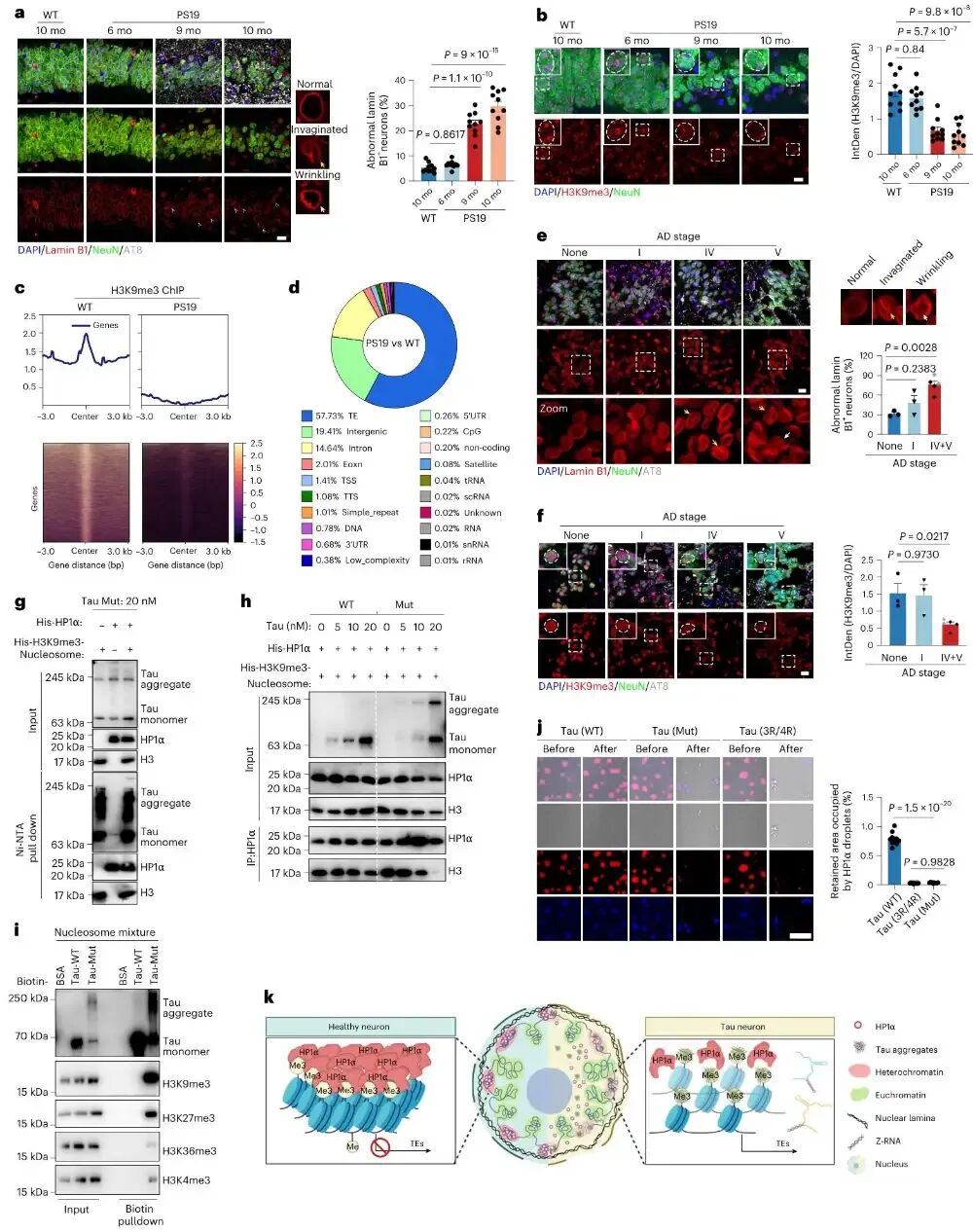
图4 tau聚集体破坏组成型异染色质压缩状态
作者从细胞、小鼠模型与人类脑组织三个层面证明,tau聚集体会特异性结合带有H3K9me3修饰的核小体,竞争性结合并驱逐HP1α蛋白,破坏异染色质的液–液相分离状态,导致核纤层结构异常、H3K9me3修饰整体下降、转座子区域表观抑制标记丢失,最终造成异染色质松散与转座子激活。
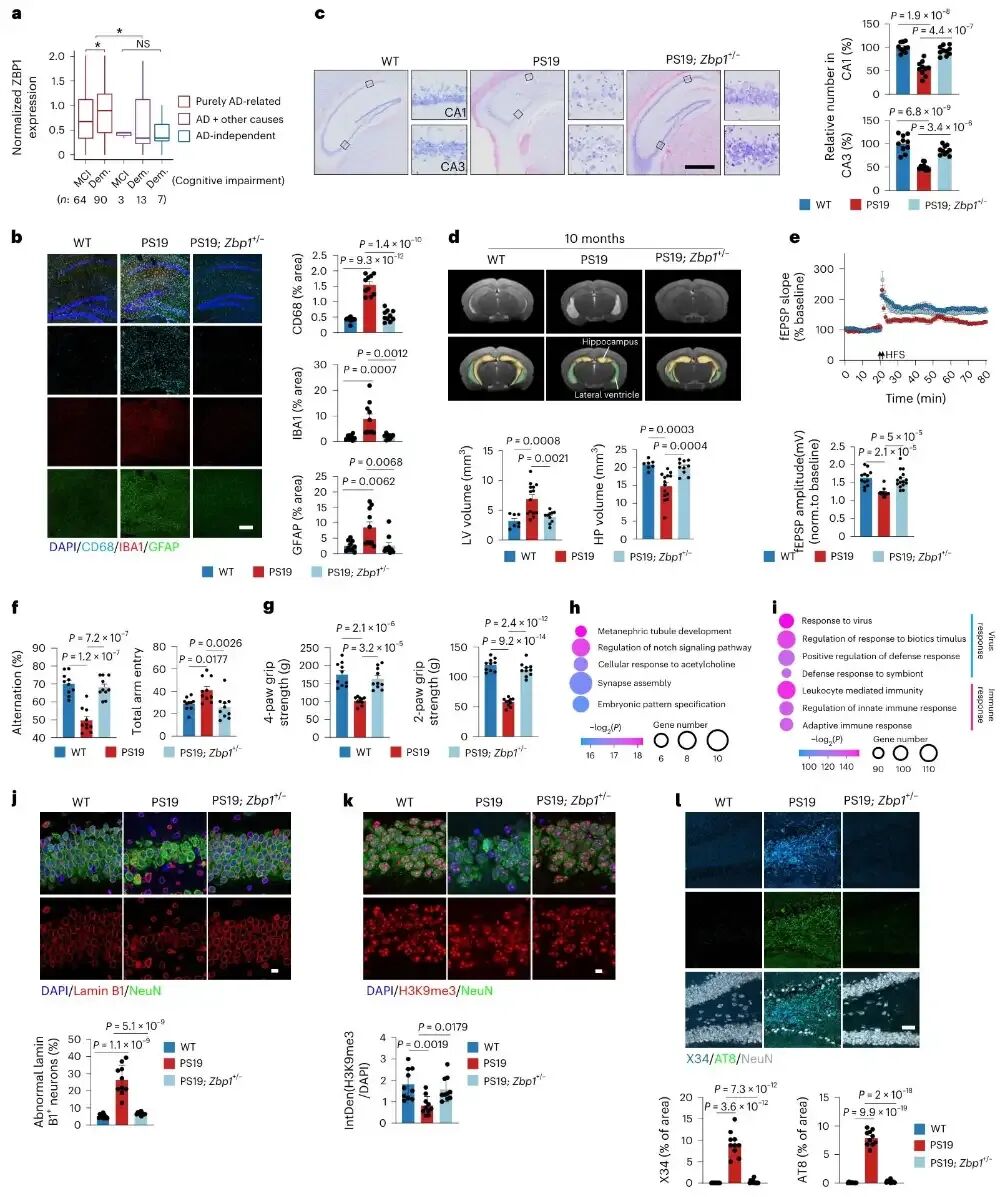
图5 Zbp1单倍剂量不足可挽救tau转基因小鼠的神经退行性表型
上图展示在10月龄PS19小鼠中,仅让Zbp1基因表达减半就能显著抑制神经炎症、保护海马神经元、恢复脑体积、修复突触可塑性并完全挽救认知与运动缺陷,同时恢复核纤层结构与H3K9me3水平、逆转转座子激活并减少tau聚集体积累。
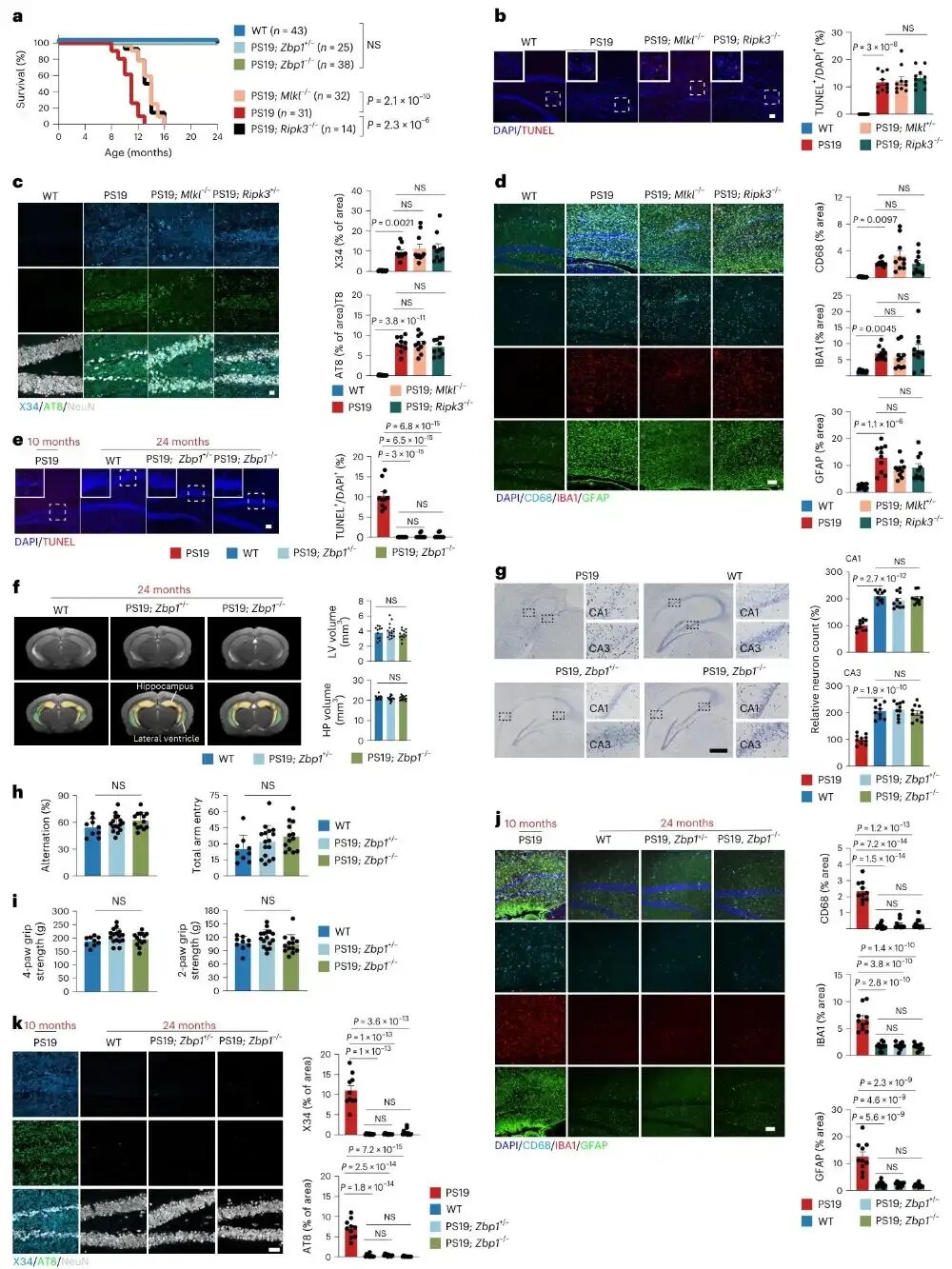
图6 靶向ZBP1可对tau病实现长期神经保护
作者通过生存分析与长期表型追踪显示,抑制下游MLKL或RIPK3仅能短期缓解神经损伤,小鼠最终仍会出现神经元死亡与tau病理复发,而敲除或半敲除Zbp1可让PS19小鼠存活至24个月且无死亡与瘫痪,完全避免脑萎缩、神经元死亡、神经炎症与tau聚集。
【全文总结】
本研究发现tau聚集体通过破坏神经元异染色质,激活转座子产生Z-RNA,进而激活ZBP1介导神经元程序性坏死,引发神经退行性病变。抑制ZBP1可显著减轻tau病理、阻止神经元死亡并长期改善认知与运动功能,为阿尔茨海默病等tau蛋白病提供了新的治疗靶点。
原文链接:
https://doi.org/10.1038/s41593-026-02299-9