深度解析医学证据,DeepEvidence为你支撑决策
淋巴瘤治疗的新进展延长了患者生存期,但也暴露了传统研究终点在疗效评估方面的局限性。《Seminars in Hematology》近日发表综述,作者提出两个基于MRD的终点,以加速药物开发和改进监管决策:改良的无进展生存期(mPFS)和MRD不可检测(uMRD)率。mPFS无事件生存期(EFS)定义为从研究入组到治疗结束时MRD阳性、临床进展或死亡的时间,具有作为传统批准的验证性替代终点的潜力;预设时间点的uMRD率可衡量缓解深度,用于早期和后线研究,以支持加速批准。
作者首先阐述了现有评估方法的潜在局限性:其可能进展缓慢、依赖不一致的影像学研究,且对分子层面的疾病负荷不敏感,而新的替代试验终点可以扩大并加速创新疗法惠及患者。技术进步已使得通过外周血实现超灵敏的MRD检测方法成为可能,包括循环肿瘤DNA(ctDNA)和免疫球蛋白克隆型测序。MRD方法已在包括大B细胞淋巴瘤、套细胞淋巴瘤、霍奇金淋巴瘤和滤泡性淋巴瘤在内的多种淋巴瘤中显示出明确的生物学原理和预后能力。将MRD纳入试验终点可以缩短试验时间,或许还能提供更准确的治疗终点。
本文详述了整合MRD的终点、详细阐述当前淋巴瘤中的MRD数据,并提供了一份关于基于MRD的替代终点用于加速药物开发的实用路线图。

引言
淋巴瘤是一组生物学和临床上多样化的血液系统恶性肿瘤,其自然史和预期结局存在显著差异。例如,霍奇金淋巴瘤(HL)最常发生于年轻人,目前通过一线治疗具有很高的可治愈性,因此在保持早期疗效的同时最小化长期毒性仍然至关重要。相比之下,在非霍奇金淋巴瘤(NHL)中,侵袭性淋巴瘤如弥漫大B细胞淋巴瘤(DLBCL)通常表现为淋巴结或其他器官的症状性疾病,如果不治疗通常是致命的,尽管通过目前的初始化学免疫治疗或二线嵌合抗原受体(CAR)T细胞疗法有可能治愈。惰性NHL,如滤泡性淋巴瘤(FL),病程呈慢性、复发性,生存期延长且有有效疗法,但如今大多数仍被认为不可治愈。另一种NHL亚型为套细胞淋巴瘤(MCL),横跨这些模式,具有异质性的行为和治疗反应,并且往往有更多的循环系统疾病。尽管近年来已开发出几种新疗法,但创新通常受限于漫长的试验时间。幸运的是,这一障碍可以通过新的替代终点来解决。在此,作者提出了包含可测量残留病(MRD)的新终点,并详细阐述了通往验证和监管应用的路线图。
当前淋巴瘤中的终点使用情况
淋巴瘤药物研发近年来取得了一些显著成功,然而那些最终改善患者预后的药物从研发到上市的时间仍然极为漫长。在淋巴瘤领域,从2006年到2024年,近2000项2期和3期试验招募了超过10万名患者,中位试验持续时间分别为44个月和49个月。只有约10%的研究性药物成功从2期过渡到3期,而不到50%的3期研究最终获得监管批准。将新药推进到一线(它们更有可能带来长期治愈)的成功率甚至更低。以DLBCL为例,在CHOP化疗方案中加入利妥昔单抗(R-CHOP)于21世纪初首次获批。在接下来的20年里,超过20项新药的一线随机对照试验(RCT)——大多数依赖于“R-CHOP + X”的附加设计——在招募了数万名患者后完成,旨在改善DLBCL的结局,但很少取得成功。展望未来,截至2025年,仅针对一线DLBCL就有至少15项额外的RCT正在进行中,更不用说其他淋巴瘤和其他线别的治疗了。加快淋巴瘤药物研发的步伐对于进一步改善患者预后似乎是必要的;能够提供更早试验结果的中间终点可以帮助我们实现这一目标。
临床试验终点不仅指导监管机构的决策,也指导临床医生和患者的决策,因此需要根据每个试验的目标进行谨慎选择。任何治疗的最终目标都是在权衡治疗获益与安全性带来的获益-风险评估时,改善患者生命的数量或质量。理想的试验终点,如总生存期(OS)或生活质量(QOL),能直接显示对患者的临床获益,包含有关疗效和安全性的关键信息。幸运的是,许多现代疗法已延长了肿瘤学领域的生存时间,然而这反过来增加了证明OS获益所需的时间和资源,从而延迟了有效疗法惠及仍有显著未满足需求的患者。对于大多数淋巴瘤(中位年龄超过60岁),预期的长OS轨迹实际上可能限制了该终点在确定药物疗效方面的实用性,并可能使某些试验不可行。特别是对于惰性淋巴瘤,许多患者现在可以通过治疗获得正常的预期寿命,并且相当一部分患者可以观察等待。尽管如此,对于许多患者而言,新的试验和治疗仍然是必要的。有效的方案后治疗也会影响OS在疗效评估中的效用。
为了解决OS作为终点的这些局限性,FDA于1992年制定了加速批准计划,允许基于替代终点更早地批准治疗严重疾病且存在未满足医疗需求的药物。事实上,现在大多数肿瘤药物都是通过这种加速途径,使用替代终点获得批准的,这些替代终点可以预测但可能不直接衡量对患者的临床获益。替代终点可以更容易、更早或成本更低地进行测量,并且已知或合理地可能预测临床获益,但不是临床获益的直接衡量标准。获得加速批准的药物必须完成一项验证性临床试验,使用能直接证明或作为真实临床获益强有力替代指标的终点。为了在传统批准的监管决策中使用,替代终点需要验证,包括来自流行病学研究和临床试验的大量、有力的证据,证明其因果性、合理性、特异性、比例性和普遍性。
在淋巴瘤临床试验中用作替代终点的各种中间终点包括总缓解率(ORR)、完全缓解率(CRR)、缓解持续时间(DOR),以及各种时间-事件指标,包括无进展生存期(PFS,捕捉至进展或死亡的时间)、无事件生存期(EFS,反映各种事件类型)、至下次治疗时间(TTNT)以及在特定时间点的疾病状态(例如惰性淋巴瘤中24个月疾病进展[POD24]或30个月完全缓解[CR30])(表1)。
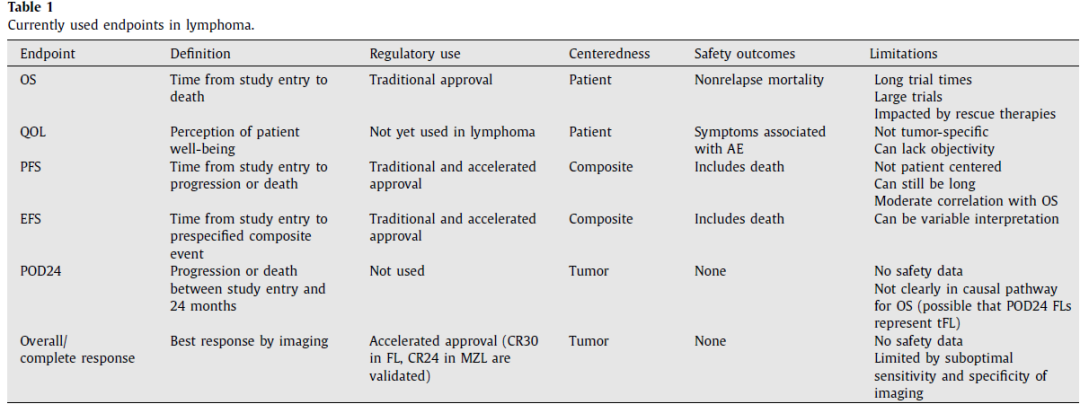
尽管未被验证为替代终点,但一种改良形式的PFS(mPFS)也用于支持HL的传统批准,其中将导致放疗的PET阳性病灶定义为一个事件,并且相应的OS后来也成功得到证实。自2013年以来,大多数淋巴瘤药物的获批是通过加速途径,使用总缓解率(ORR)和/或缓解持续时间作为主要终点(图1A-B)。然而,尽管这种由针对多线复发或难治性疾病的后期治疗线别批准所驱动的普遍趋势存在,但在一线或二线治疗中,各种生存终点更常用于支持传统批准(图1C)。
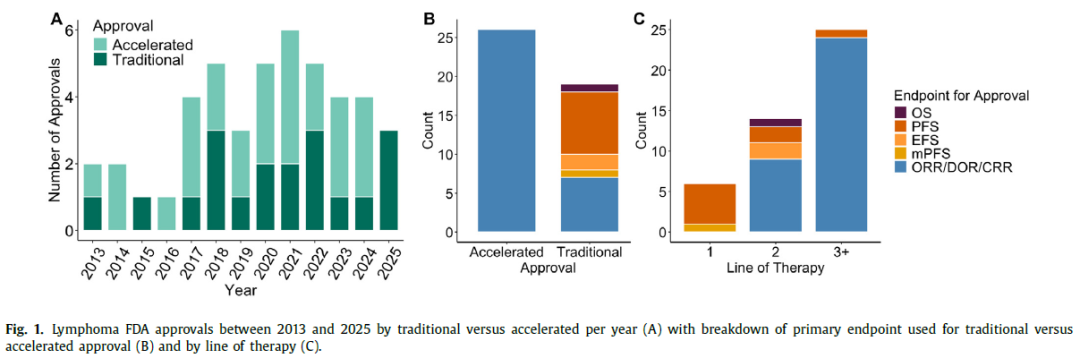
虽然经过验证的替代终点提高了淋巴瘤药物上市的速度,但仍有很大的改进空间。早期试验中对新药的疗效评估倾向于依赖总缓解率和完全缓解率,这类缓解评估通常通过PET/CT来判断,而PET/CT在特异性和敏感性方面都有固有的局限性。尽管可能有复发倾向,但大多数淋巴瘤在影像学上已经具有极高的缓解率。由于这种影像学缓解率可能只会随着新疗法的出现而进一步提高,因此需要更灵敏的残留病灶检测试验来更真实地确定治疗效果。此外,这种影像学通常不能提供分子水平上疾病负荷的准确、定量测量,限制了其在缓解深度或动力学方面的应用。改进的疾病缓解评估可以提供更准确的治疗效果图景,从而更好地对试验和药物批准进行优先排序。
PFS和EFS常用于淋巴瘤,特别是在早线治疗中,其目标是更快速、更准确地评估治疗效果。然而这些终点通常仍需要很多年才能完成试验以等待进展事件,往往依赖PET/CT或医生判断(这些方法存在局限性),并且也未能捕捉到分子水平的残留病灶。此外值得注意的是,PFS和EFS作为OS替代指标的水平并不完美。
对肿瘤负荷更敏感并能测量更深缓解的早期终点具有许多潜在优势。今天仍有太多患者死于淋巴瘤,因此更有效的新疗法是一个紧迫的未满足需求。使用替代终点的中间试验终点有望通过更快的试验结果和更小的试验规模来加速药物开发,意味着药物可以在多个阶段试验中开发和测试,成败能更早确定,从而使成功的策略能够更快地获得潜在的监管批准。对于药物开发者来说,这种时间效率也意味着显著的成本节约,使试验更具可行性。理想情况下,将转化为丰富的可用临床试验,从而为医疗系统带来更多药物和更便宜的治疗方法。
纳入MRD能否克服当前终点的局限性?
MRD有潜力解决淋巴瘤当前终点的若干局限性。MRD的可操作应用已在其他血液恶性肿瘤(包括急性淋巴细胞白血病、慢性粒细胞白血病,以及最近在多发性骨髓瘤中)得到充分证实,然而淋巴瘤领域相对滞后。近年来,重大的技术进步使得利用血液样本能够以高灵敏度更准确地检测残留淋巴瘤。早期方法侧重于使用PCR和FISH对完整的循环细胞进行检测,但由于其特异性和灵敏度不理想(尤其对于非白血病性疾病),它们在MRD检测中的应用受到限制。二代测序(NGS)可使用多种方法,包括通过免疫球蛋白高通量测序(IgHTS)检测免疫球蛋白(Ig)克隆型,或通过CAPPseq和PhasED-seq等方法检测无细胞循环肿瘤DNA(ctDNA),导致淋巴瘤MRD检测的准确性得到了显著提高。MRD方法具有不同的特征,包括检测方法、检测下限(LoD)、检测的区室、基因分型能力和临床表现,这些已在专门的综述中总结过。协调样本采集和标准化性能指标将是优化其作为终点用途的关键(图2)。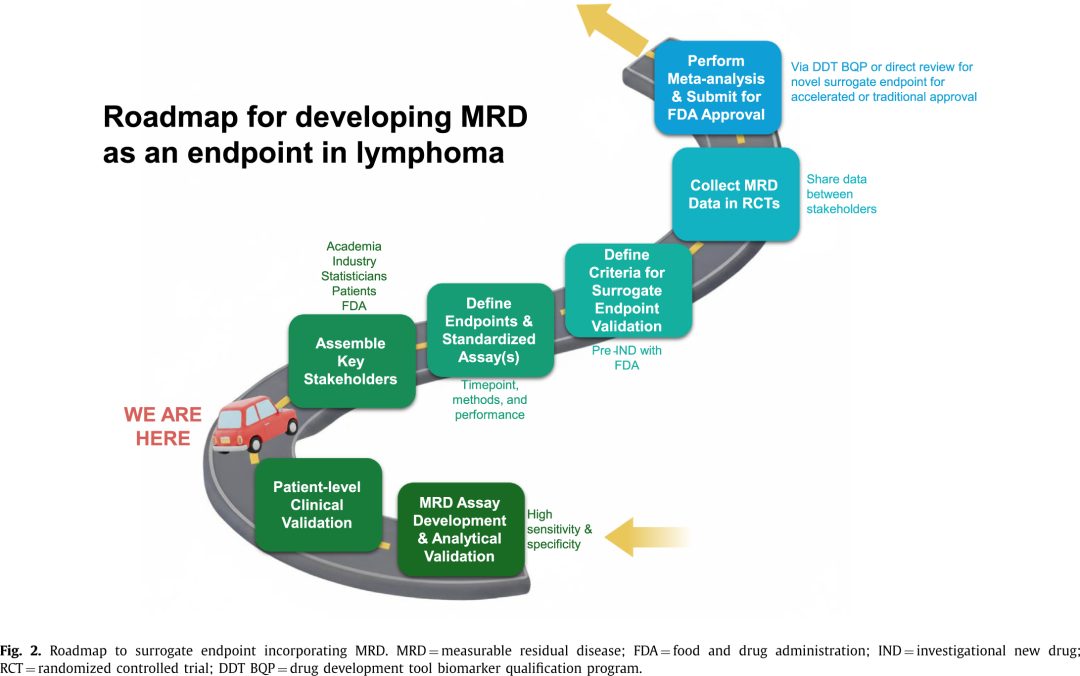
将MRD作为替代终点的监管考虑
2020年,FDA发布了关于在血液恶性肿瘤药物和生物制品开发中使用MRD的监管考虑指南。2024年,FDA发布了类似指南,涉及在实体瘤中使用MRD,下文将进一步描述。不幸的是,淋巴瘤被排除在这两个指南框架之外,可能是由于当时缺乏足够灵敏度的MRD检测方法,尽管相同的原则应该适用。FDA有两种机制支持新型替代终点的批准,详见下文。值得注意的是,这两种机制都需要证明生物学合理性、证明对传统临床结局具有预后价值的流行病学证据,以及证明治疗效果与预期临床结局相对应的临床试验证据(图2)。
第一种是通过药物开发工具 (DDT) 认证过程,特别是生物标志物认证程序 (BQP)。其目的是对滴滴涕的特定使用环境进行认证,以便其解释和应用在药物开发和监管申请审查中可靠。然后,DDT可以作为研究用新药 (IND) 申请、新药申请 (NDA) 或生物制品许可申请 (BLA) 的一部分纳入,而不必重新考虑生物标志物。FDA 指出,鉴定生物标志物需要强有力的科学证据,使用更高的证据标准作为替代终点。
第二种是通过药物评价与研究中心(CDER)和生物制品评价与研究中心(CBER)的审评部门批准使用新型替代终点。对于此过程,根据治疗类别,申办方或团体与相应的FDA审评部门会面,为特定治疗适应症提交支持拟议替代终点的数据。这些支持数据可以来自以前的临床试验、针对该疾病领域多项试验的荟萃分析,或其他支持使用拟议终点的数据(图2)。在FL中,CR30遵循了这一途径。在此,一个合理地可能预测临床获益的替代终点可以支持加速批准(中间终点),而一个经过验证的替代终点可以支持传统批准。这两种类型替代终点的区别在于支持拟议终点的临床数据的质量和数量。
2024年,FDA发布了关于在实体瘤药物开发中使用ctDNA的指南,强调了使用ctDNA动力学或清除作为药物反应的衡量标准,以在早期试验中了解药物活性,并在根治性治疗中作为早期终点。前者对惰性淋巴瘤尤其实用,而后者则定义了在侵袭性淋巴瘤中的应用。
要想使用 ctDNA 作为早期终点,必须满足几个标准,包括来自前瞻性随机临床试验的数据(包括评定 ctDNA 终点),在治疗前后收集 ctDNA 以表征 ctDNA 减少或清除与长期结果的相关性的试验,多个时间点的定量 ctDNA 分析,并最终进行荟萃分析来验证 ctDNA MRD 检测的使用(图2)。重要的是,MRD检测必须可靠,并达到足够的临床灵敏度和特异性,以支持预期用途。MRD 采样的时间和质量控制也应一致。
纳入MRD的潜在终点
MRD不可检测(uMRD)率
将MRD用作终点的最简单方法是作为治疗后特定时间点MRD不可检测到率或MRD清除率的衡量指标(表2)。

遵循多发性骨髓瘤的模式,在肿瘤药物咨询委员会(ODAC)以12-0的投票结果一致推荐批准其用于监管审批后,FDA批准将LoD低于10-5的检测方法所得的uMRD用作替代终点。在淋巴瘤中,许多早期临床试验,特别是针对晚期线别治疗的试验,其主要终点依赖于使用影像学测量残留病灶的总缓解率或完全缓解率(图1),但这种影像学的灵敏度通常不理想。众所周知,淋巴瘤对治疗反应良好,通常在影像学上表现为部分或完全缓解,但许多此类缓解并不持久。此外,阳性的PET/CT可能反映残留的炎症活动,而非持续的淋巴瘤,因为18F-FDG摄取并非肿瘤特异性。在各类淋巴瘤中,MRD检测能更好地捕捉缓解深度,通过uMRD率或MRD清除率来衡量,这是单纯影像学无法看到的,因此通过MRD进行的缓解深度评估可以提供更真实的治疗反应衡量标准。
对于目前基本不可治愈的惰性淋巴瘤,这种早期终点尤其有用,因为等待PFS或OS事件发生的试验时间可能极长,需要大量的随访时间才能成熟。此外,FL患者通常以较低的肿瘤负荷和对治疗的反应性为特征。在各种研究和疗法中,达到uMRD的FL患者具有更好的PFS和OS。在早期试验或后期治疗线别中,使用uMRD可以通过更早地识别最有效的药物来加速药物开发。
MCL的疗法现在包括作为维持治疗或无限期治疗的靶向药物。uMRD率可用于理解治疗不同阶段的疗效。对于可治愈的侵袭性淋巴瘤,治疗后(尤其是在早期线别中)的uMRD率代表了高治愈可能性。在后期线别中,它代表了超越影像学的一种有用的药物疗效衡量指标。
在单一治疗结束时间点的uMRD率作为监管试验终点确实存在局限性(表2)。与总缓解率或完全缓解率非常相似,uMRD率不包括任何安全性数据,更早的试验结果可能掩盖重要的迟发性毒性信号。不良事件数据需要单独分析,并根据不同治疗间uMRD率的获益进行权衡。如前所述,MRD检出率可能受检测方法的LoD和检测区室的影响,因此需要协调统一使用检测方法。此外,测量的时间点会影响MRD率和至进展时间,而缓解持续时间则被忽略。
改良的无进展生存期(mPFS)
考虑到治疗结束时MRD不可检测的局限性,改良的无进展生存期(mPFS)可能是一个理想的替代终点,值得验证(表2)。在ECHELON-1研究中,该研究促成了BV-AVD获得FDA批准,mPFS事件被定义为标准PFS事件(治疗开始后任何时间的HL复发或进展或任何原因导致的死亡),以及需要RT的持续性PET阳性病灶(表明原发化疗失败)。鉴于MRD阳性具有极高的阳性预测值,建议在mPFS终点中纳入MRD,以捕捉至事件结局的时间,其中除了常规的PFS事件(包括治疗开始后任何时间的淋巴瘤临床复发或进展、任何原因导致的死亡)外,事件应包括在化学免疫治疗完成后一次或多次MRD检测阳性(表明存在残留病灶)(表2)。
尽管mPFS在许多情况下可能有潜在获益,但该终点的初始应用是在DLBCL或HL等淋巴瘤的根治性治疗中。学者在一组接受一线化学免疫治疗并通过PhasED-seq评估治疗结束(EOT)MRD的DLBCL患者中,为mPFS建立了概念验证(图3A-C)。对于预期的25%事件发生率(如在最近的POLARIX等DLBCL试验中所见),mPFS可以将试验结果读出时间缩短一年。重要的是,PFS和mPFS事件之间的一致性很高(94%),不一致的事件可能是由于PFS随访时间较短所致。高IPI评分仍然是mPFS预后不良的预测因素,如同对PFS和OS一样,表明风险分层对这一新终点同样适用。
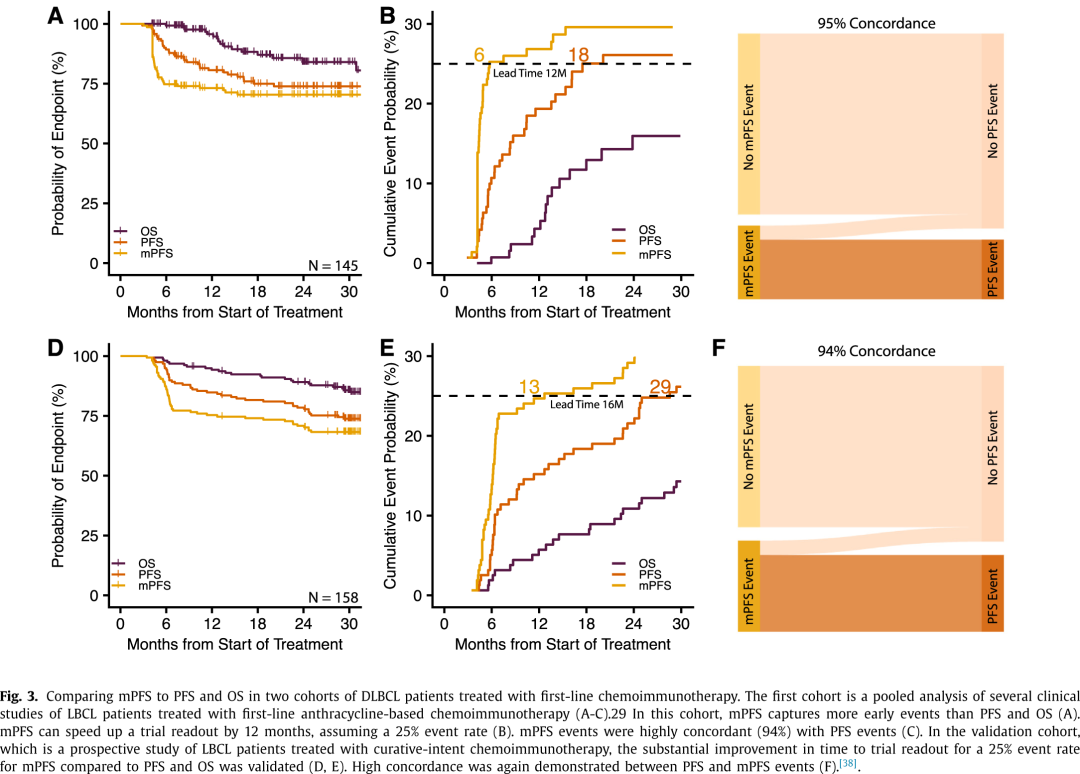
验证mPFS作为替代终点需要标准化一个高度特异和灵敏的MRD检测方法(表2)。灵敏度较低、LOD较高(如10-4)的MRD检测方法并不能显著缩短试验结果读出时间。同时,特异性较低的检测方法会妨碍mPFS事件的准确报告。与MRD检测不同,mPFS确实纳入了安全性数据,包括将死亡作为终点。然而更早的结果读出可能限制对长期临床获益或毒性的理解,需要继续研究或加强上市后监测(表2)。荷兰一组接受根治性一线化学免疫治疗的新诊断LBCL患者队列中验证了该发现(图3D-F)。结果高度一致,试验结果读出时间显著缩短,且mPFS与PFS事件之间具有高度一致性(图3C和F)。
特定时间点的uMRD率可以作为早期试验或后期治疗线别中的一个重要的中间终点;然而纳入时间-事件和安全性数据使得mPFS成为更值得进行验证的替代终点(表2)。将MRD整合为淋巴瘤终点的另一条途径是通过基于MRD结果指导治疗的阳性风险适应性试验。先前在其他癌症中的努力表明,当后续治疗的选择基于MRD状态并成为新的治疗标准(例如,用于MRD+ ALL的贝林妥欧单抗和CML中的二线TKI)时,MRD往往会成为先前治疗线疗效的适当终点。事实上,同样的模式也可能扩展到淋巴瘤,取决于目前正在进行的DLBCL和HL中几项风险适应性试验的新兴数据。
淋巴瘤中支持MRD生物学合理性和可操作性的数据是什么?
大B细胞淋巴瘤
大B细胞淋巴瘤(LBCL)是一组异质性的侵袭性淋巴瘤,包括DLBCL、高级别B细胞淋巴瘤(HGBCL)和原发性纵隔B细胞淋巴瘤(PMBCL),通过标准的初始化学免疫治疗以及二线CAR-T细胞治疗可以治愈。治疗后检测到残留的LBCL表明治疗无效且预后不良,这种残留病灶通常通过在治疗结束时的PET/CT进行测量。大多数LBCL与实体瘤更相似,通常缺乏丰富的循环肿瘤细胞,限制了外周血流式细胞术或其他细胞技术在MRD检测中的应用。尽管如此,大多数LBCL具有相对较高的由淋巴瘤细胞释放的循环肿瘤DNA。目前已有多个队列评估了克隆型测序和杂交捕获测序在LBCL初始化学免疫治疗结束时检测MRD的应用。
初步研究表明,血浆中的克隆型测序能够在临床复发前持续识别出血浆中的复发性疾病。然而,这些数据以及一项关于克隆型测序用于DLBCL监测的前瞻性验证显示,其在治疗后检测疾病的灵敏度和特异性均不理想。特异性的局限性可能源于未能正确识别优势克隆型,或者识别出正常B细胞共享的低多样性正常轻链克隆型。血浆中鉴定的克隆型数量较少限制了对低水平疾病的检测灵敏度,尤其是在后期时间点。一种血浆克隆型测序方法clonoSEQ已获得Medicare批准用于DLBCL。最近,用于DLBCL的clonoSEQ更新了检测方法,允许从10mL血浆中提取ctDNA,旨在提高临床灵敏度。
下一代DLBCL MRD检测方法是围绕通过杂交靶向NGS检测肿瘤特异性无细胞DNA分子(ctDNA)而开发的。CAPP-Seq 专注于超高深度测序淋巴瘤基因组的复发突变区域。虽然CAPP-seq在治疗前肿瘤负荷较高时进行预处理基因分型和早期分子反应(EMR)动力学方面表现出色,但由于检测单核苷酸变异(SNV)时固有的背景错误率(1/10,000),它在后期时间点的灵敏度也受到限制。值得注意的是,ctDNA动力学(如EMR)在LBCL中具有很高的预后价值,也可能作为终点具有价值。
这些局限性促使了PhasED-seq的开发,其通过评估同一DNA链上多个SNV的同期变异(即相联变异)来降低背景错误并提高检测下限]。由于活化诱导胞苷脱氨酶(AID)的活性导致在基因组的特定区域出现紧密聚集的突变,因此相联变异在B细胞NHL中很常见。最近的两个队列证明了PhasED-seq在DLBCL中用于评估治疗结束(EOT)MRD的实用性。在一个汇总队列中,标准化学免疫治疗结束时PhasED-seq检测阳性的结果具有更好的灵敏度、特异性、阳性预测值和阴性预测值。PhasED-seq在EOT的灵敏度几乎是LOD为10^-4的检测方法(如CAPP-seq)的两倍。EOT PhasED-seq检测对预测患者结局的高准确性在前瞻性HOVON国家队列中得到了验证。在两个队列中,PhasED-seq MRD检测对结局的预测能力均显著优于目前用于确定疾病反应或进展的PET/CT。目前有多项一线试验正在LBCL中进行基于MRD的风险适应性方法研究,包括剂量递增方法(如ALPHA3和SAKK38/19),以及剂量递减方法(包括SHORTEN-ctDNA)。
在二线治疗中,CAR-T细胞疗法已成为一种根治性治疗。对于CAR-T细胞疗法,使用clonoSEQ、CAPP-seq和PhasED-seq在多个时间点检测输注后的MRD均具有预后价值。在所有三项研究中,MRD检测都能在复发前发现疾病,并且在第28天或4周之后进行的评估比当时进行的评估更具预后价值。在TRANSFORM试验中,与标准治疗组相比,liso-cel组的ctDNA清除与改善的EFS相关。
霍奇金淋巴瘤
经典型HL是一种独特形式的淋巴瘤,具有双峰年龄分布,大多数发生在年轻患者中,在老年人中有一个较小的峰值,通过化学免疫治疗具有很高的治愈率。HL的特征是存在Hodgkin/Reed-Sternberg细胞,DNA这些细胞在组织中很少,限制了肿瘤标本的分子分析。相比之下,HL患者的血浆异常富含ctDNA,为基因组特征描述、量化肿瘤负荷和确定MRD提供了一个有价值的工具。两项包括初治和复发/难治性HL的多中心研究通过CAPP-seq检测的MRD,显示出在中期和治疗结束时间点均具有很高的预后价值,为PET/CT提供了额外的阳性预测价值。然而CAPP-seq的NPV与PET/CT一致,表明通过更低的LOD可能还有改进空间。PhasED-seq在一组年龄偏大的HL患者中进行了治疗期间和一线治疗后的评估,在中期和治疗结束时间点,PhasED-seq对PFS和OS均具有很高的预后价值。当控制PET/CT上的EOT CR时,通过PhasED-seq评估的EOT MRD仍具有预后价值,表明其具有超越影像学的额外价值。PhasED-seq评估的EOT MRD还在一项单臂帕博利珠单抗 + AVD试验中进行了评估,结果显示在EOT时达到uMRD的患者百分比高于PET/CT,并且能更好地预测唯一的复发事件,突显了PhasED-seq的灵敏度和特异性,以及在添加检查点抑制剂(现已成为标准治疗)后PET/CT的局限性。PhasED-seq目前正在PRECISE-HL研究中进行评估,以限制对接受4个周期纳武利尤单抗 + AVD治疗后达到uMRD的患者进行化疗。
套细胞淋巴瘤
MCL是一种异质性的淋巴瘤,其特征为t(11,14)易位,使CCND1基因处于IGH基因的控制之下。与前述其他淋巴瘤不同,MCL经常表现出循环肿瘤细胞。因此,多种MRD检测方法具有实用性,并在过去十年中得到了研究,一致表明EOT MRD是MCL的一个高效预测因子。
MCL中最初的MRD方法包括多参数流式细胞术(MFC)和实时定量聚合酶链反应(RT-qPCR)。MFC靶向MCL的免疫表型,而RT-qPCR靶向MCL典型的IgH或t(11,14)重排,两种MRD检测都可以在血液或骨髓上进行,但对于使用这些检测方法进行MRD检测,骨髓通常优于外周血。两者在临床实践中的应用都面临障碍;MFC缺乏标准化且灵敏度有限,而RT-qPCR劳动强度大。此外,RT-qPCR仅跟踪单个分子靶标,限制了灵敏度,并且高达25%的患者无法鉴定出分子标志物。
二代测序可以通过对大量基因组改变(包括免疫球蛋白位点的重排以及SNV和肿瘤特异性突变)提供大量读数来克服许多这些限制。clonoSEQ通过测序单核细胞实现这一点,并报告在骨髓和外周血中对MCL均具有高灵敏度和特异性的1/1,000,000的LOD。在EA4151试验中,诱导化学免疫治疗后clonoSEQ检测不可检测,且影像学和实验室检查显示完全缓解的患者,并未从在利妥昔单抗维持治疗的基础上添加自体干细胞移植中获益。clonoSEQ还可用于理解对治疗方案的缓解深度,如在ECHO试验中,与BR加安慰剂相比,在苯达莫司汀和利妥昔单抗(BR)基础上添加阿卡替尼的患者具有更高的uMRD率,并且PFS存在相应差异。clonoSEQ目前正被用于研究在接受三联靶向治疗的MCL患者中,基于MRD的风险适应性方法以停止持续治疗。
滤泡性淋巴瘤
滤泡性淋巴瘤是最常见的惰性NHL,其特征通常是正常寿命,但疾病自然史多样,常需要多种治疗。其特征是85%的患者存在t(14;18)易位,使BCL2基因处于IGH调控区的控制之下,导致抗凋亡BCL2的过表达。早期的MRD检测方法使用RT-qPCR在骨髓和血浆中量化这种易位。自从在FL的化疗中加入利妥昔单抗以来,通过外周血RT-qPCR检测的uMRD率通常接近90%,在骨髓中略低。RT-qPCR检测的MRD在预测缓解深度方面具有实用性,例如在III期GALLIUM研究中。在该研究中,奥妥珠单抗+化疗和利妥昔单抗+化疗之间的临床缓解率相似,但奥妥珠单抗组的PFS有所改善。这很可能是由于奥妥珠单抗+化疗组观察到的缓解率更高,其中92%达到了uMRD,而利妥昔单抗+化疗组则较低。
尽管MRD检测具有预后价值,但对于一种不可治愈的疾病,高uMRD率表明检测方法需要比RT-qPCR有更高的灵敏度。因此,ctDNA在FL中用于MRD的作用一直在研究中。与侵袭性淋巴瘤相比,FL患者在基线时倾向于具有较低的ctDNA,进一步突显了对超灵敏MRD检测方法的需求。通过靶向测序进行ctDNA评估对于FL的疾病监测是有用的,但其在EOT MRD评估中的应用与预后相关,尽管在小队列中结果有些混杂。具有更低检测下限的PhasED-seq可以在治疗期间和治疗后以较低水平检测FL,并具有高预后能力。
要将MRD纳入支持加速或传统批准的替代终点,还需要哪些数据?
目前关于MRD的累积数据已可靠地证明其生物学合理性,且在患者水平上对PFS和OS具有强大的预后价值。这些观察结果在各个淋巴瘤亚型、治疗方法和时间点之间普遍一致,为替代终点所需的前两个标准提供了理由。在不同淋巴瘤类型和治疗背景下使用一致检测方法进行的进一步研究,对于建立针对组织学和治疗的、患者水平上的全局风险比至关重要。全面确立患者水平的预后价值意味着mPFS合理地可能预测临床获益,因此可用于加速批准。建立一个经过验证的替代终点需要试验水平的关联性,证明其能可靠预测临床获益。2024年,FDA批准MRD作为多发性骨髓瘤中加速批准的监管决策的早期终点。这一跨越15年的里程碑式成就,仍然是MRD获准用作终点的唯一适应症。通过在未知的监管领域开辟道路,它为淋巴瘤学界提供了一份蓝图,可以在此基础上高效地批准一个包含MRD的早期终点,以加快药物开发[58]。为了实现这一壮举并追随多发性骨髓瘤的脚步,关键学术界、工业界和监管机构之间的标准化与合作至关重要(图2)。
试验水平的关联性是指治疗对替代终点(如mPFS)的效果与对真实临床终点(PFS或OS)的效果之间的相关性,评估这一点需要多个同时测量mPFS和真实临床终点的随机临床试验。理想情况下,这些RCT应涵盖不同的淋巴瘤类型、治疗方法和时间点,以理解该终点的普适性。阳性和阴性试验都必须包括在内。对于像DLBCL这样的疾病,一线试验很少取得阳性结果,这可能是一个挑战,尽管后期治疗线别和其他淋巴瘤可能提供这些数据。
个体水平的全局风险比和试验水平的治疗效应可以通过荟萃分析进行评估。为了高效完成这一荟萃分析所涉及的庞大任务,应与关键利益相关方举行初步会议,包括监管机构、淋巴瘤试验专家和科学家、统计学家、生物技术和制药工业界、基金会以及患者代表(图2)。在荟萃分析中确立mPFS作为PFS或OS的替代指标的标准必须预先指定,包括用于患者水平相关性的全局风险比和用于试验水平相关性的R2方法。提供满足这些标准的稳健统计估计将需要足够数量的包含EOT MRD数据的试验。这需要学术界和工业界资助的试验中的大规模合作努力,随后进行数据共享(图2)。
由于检测方法性能和时机可能影响mPFS和uMRD率,因此跨试验的标准化和协调统一是关键之一,并需要利益相关方达成共识(图2)。基于在DLBCL中的分析,检测下限为百万分之一的检测方法可维持EOT时间点所需的高临床灵敏度和特异性。在基线ctDNA水平较低的惰性淋巴瘤中,来自更低LOD的更高灵敏度对于MRD评估更为重要。在HL中,CAPP-seq和PhasED-seq均已显示出显著效用,而后者可能带来更高的灵敏度,可能会改善NPV。在MCL中,外周血中百万分之一的LOD与骨髓活检的表现相似,允许进行创伤较小的监测。
基于这些数据的一致性,建议使用检测下限或分析灵敏度为百万分之一的检测方法。可以使用tumor-informed或tumor-naïve检测方法,只要它们经过充分验证并能达到百万分之一的分析LOD。目前,只有tumor-informed MRD检测方法能在淋巴瘤中达到此LOD。作者注意到,其中一位作者(AA)作为一家销售淋巴瘤MRD检测方法的公司的创始人存在利益冲突,但作者认为这一建议是有数据支持的。患者水平的分析LOD可能受检测方法的错误率、追踪的突变数量以及输入DNA的影响,因此需要一致且严格的质量控制。样本采集的最佳时机受治疗和组织学类型的影响。对于侵袭性淋巴瘤的固定疗程方案,建议在治疗完成后至少4周以及此后每6个月间隔采集一次MRD样本。对于持续治疗方案,MRD样本采集应至少每年进行一次。
结论
将MRD纳入新型替代终点可以显著加快淋巴瘤的药物开发,并高效地为有需求的患者带来更多疗法。将mPFS确立为支持传统批准的验证性替代终点,或将uMRD率确立为支持加速批准的中间替代终点,需要淋巴瘤学界广泛合作。为MRD实现稳健的个体水平和试验水平验证,将依赖于跨多个临床试验协调统一地收集MRD数据,从而能够进行荟萃分析。
关键的下一步是召集淋巴瘤MRD领域的所有关键利益相关方,包括FDA,以统一共同目标,就检测方法选择和时机达成共识,并定义资格认证替代终点的关键标准(图2)。这个淋巴瘤MRD联盟定期开会以完善这些标准并推动实施。当达成初步共识后,可以提交预IND申请,以开启正式的FDA讨论,随后制定统计分析计划。随着来自正在进行和未来试验的额外MRD数据的积累,制药和学术界申办方之间的数据共享对于完成建立个体水平和试验水平相关性的汇总分析至关重要。然后可以将对这些数据的全面评估提交给FDA进行正式的资格认证。通过大规模合作、协调统一的MRD检测、共同的数据收集和开放的数据共享,淋巴瘤学界可以产生新型替代终点所需的证据,从而加速淋巴瘤领域的创新并改善患者的预后。