深度解析医学证据,DeepEvidence为你支撑决策
睾丸性索间质肿瘤(TSCSTs)是一类罕见肿瘤,包含形态多样、呈性索、间质或混合表型的多种肿瘤。准确分类对临床管理至关重要,因为多种肿瘤类型与肿瘤易感综合征或其他基础疾病相关。此外,部分 TSCSTs 具有临床恶性行为且对全身治疗无效,因此识别具有恶性潜能的肿瘤尤为重要。以往,这类肿瘤的分类主要依赖形态学,免疫组化除少数例外情况外大多缺乏特异性。近年来,多项研究提供了新数据,深化了对这类肿瘤的认知,影响了其分类与预后评估。本文总结最具临床意义的 TSCSTs 组织学类型,重点介绍具有潜在临床应用价值的最新研究进展。


截取自WHO ( 2022) 泌尿系统和男性生殖器官肿瘤分类
研究背景
90% 以上的睾丸肿瘤起源于生殖细胞,更具体地说,起源于卵黄囊胚外中胚层的生殖细胞前体,被归类为睾丸生殖细胞肿瘤(TGCTs)。TGCTs 的发生受多种因素影响,包括易感基因变异与环境损伤,这些因素会干扰生殖细胞前体与不同发育阶段生殖细胞的正常发育,或促进其恶性转化。第二大类睾丸肿瘤的认知程度显著更低,这类肿瘤起源于(或向)睾丸性索衍生物与 / 或间质成分分化,占所有睾丸肿瘤的 4%–8%,被归入广义的睾丸性索间质肿瘤(TSCSTs)范畴。
由于睾丸肿瘤总体发病率较低(约占全球新发男性恶性肿瘤的 1%),且 TSCSTs 在其中更为罕见,大多数执业病理医师对其接触有限。因此,这类肿瘤的诊断与准确分类对风险分层和临床管理至关重要,但往往极具挑战性。诊断难点还在于不同 TSCSTs 亚型间存在显著的形态学与免疫表型重叠,部分肿瘤可能是肿瘤易感综合征的首发表现。此外,除少数例外,免疫组化所用标志物大多缺乏特异性,需结合背景解读。例如,呈性索表型的肿瘤(如支持细胞瘤)会表达体腔上皮性索衍生物的标志物(如 WT1、SOX9),但这些标志物也在纯间质肿瘤(如莱迪细胞瘤)中表达。因此,TSCSTs 的诊断解读需要经验,常需由经验丰富的泌尿病理医师进行中心会诊。
从临床角度看,最常见的 TSCSTs 组织学类型中,90%–95% 为良性,经睾丸切除术或选择性患者的保留睾丸手术即可治愈。但存在一些具有重要临床意义的例外,需诊断医师识别:肌样性腺间质肿瘤、纤维瘤 / 卵泡膜细胞瘤、幼年型颗粒细胞瘤均呈惰性生物学行为;而新发现的融合驱动型 TSCSTs 在临床管理中应一律视为恶性。恶性 TSCSTs 的治疗与随访面临诸多挑战,多数对化疗、放疗无效,手术成为唯一可能治愈的手段。这凸显了对原发肿瘤进行准确恶性风险评估的重要性,以识别需要密切随访或更积极治疗(包括考虑初始腹膜后淋巴结清扫)的患者。遗憾的是,现有评估体系均无法可靠识别所有恶性肿瘤,导致部分 TSCSTs 被归为恶性潜能未定。尽管近期分子研究发现了与侵袭性行为相关的分子变异,但尚无可跨组织学类型识别 TSCSTs 恶性潜能的通用风险分层标志物。
本文总结 TSCSTs 的临床病理特征与诊断要点,重点介绍可能优化分类与预后评估的最新研究进展。
莱迪细胞瘤
莱迪细胞瘤是最常见的 TSCSTs,占该类肿瘤的 70%。成人型约占 80%,占睾丸肿瘤的 1%–3%,典型发病年龄为 30–60 岁;儿童型约占 20%,占睾丸肿瘤的 3%–9%,发病高峰为 5–10 岁。莱迪细胞瘤多为散发性,偶可作为遗传性平滑肌瘤病肾细胞癌综合征或克氏综合征的表现之一。与克氏综合征相关的肿瘤通常为莱迪细胞增生背景上的优势结节。临床表现在青春期前与青春期后患者存在差异,部分原因是性激素产生的影响不同。成人多表现为单侧可触及肿块或局部模糊症状;性激素(包括雌激素,可直接分泌或由睾酮外周芳香化产生)分泌可导致男性乳房发育。儿童性激素分泌常导致同性性早熟。
驱动莱迪细胞瘤的致癌事件尚未完全阐明。儿童病例似乎总是与绒毛膜促性腺激素 / 黄体生成素受体(LHCGR)p.D578H 功能获得性变异相关。成人肿瘤多为 LHCGR 野生型,一小部分但具有临床意义的亚群存在延胡索酸水合酶(FH)功能缺失性变异。偶见携带 GNAS 热点突变的莱迪细胞瘤,但这并非高度频发事件。成人莱迪细胞瘤中最常见的频发性基因组变异为 CTNNB1 功能获得性变异,在该肿瘤类型中主要表现为亚克隆事件。
大体上,典型的莱迪细胞瘤为直径小于 5cm、边界清晰的实性小结节,切面呈均匀的黄褐至金棕色。组织学上(图1),多数肿瘤由片状、巢状分布的大多边形细胞构成,胞质丰富嗜酸性颗粒状,细胞核圆形、轮廓规则,可见单个明显核仁。散在的毛玻璃样染色质细胞是有用的诊断线索。尽管存在形态变异,多数肿瘤无提示性索分化的生长方式,如规整小管或条索结构。杆状胞质嗜酸性雷尼克结晶具有诊断特异性,但临床实践中仅见于不超过三分之一的莱迪细胞瘤。部分肿瘤可见胞质内或细胞间透明小球,但无特异性,支持细胞瘤中也可出现。脂褐素色素常见,可能是莱迪细胞活跃胆固醇代谢的结果。肿瘤间质通常稀少且不明显,可见丰富的纤细分支毛细血管网。莱迪细胞瘤对一般性索间质标志物 SF‑1 和抑制素呈阳性,两者灵敏度均超过 90%。体腔上皮衍生物(包括性索)特征性转录因子如 WT1、SOX9 和 FOXL2 多为阴性,仅见于较小比例病例。
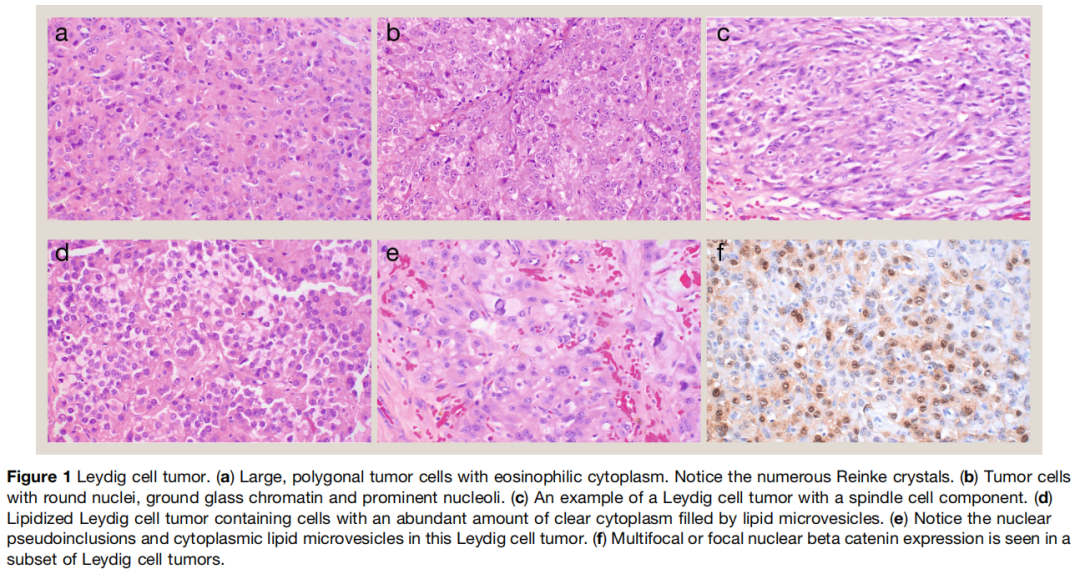
图1
多种不常见形态变异可导致诊断陷阱。少见细胞形态特征包括脂质化肿瘤细胞(胞质完全充满脂质微泡)、梭形细胞和多核肿瘤细胞。此外,部分莱迪细胞瘤可出现更丰富的不同程度玻璃样变或水肿性间质,以及微囊结构。莱迪细胞瘤的微囊型结构易被误诊为卵黄囊瘤。部分肿瘤可出现脂肪化生灶,识别该现象对排除门部脂肪组织受累至关重要,后者是莱迪细胞瘤恶性行为的危险因素,也是肾上腺生殖综合征睾丸肿瘤(TTAS)的常见表现。罕见情况下,莱迪细胞瘤可出现散在砂粒体样钙化,易被误诊为大细胞钙化性支持细胞瘤。但大的分叶状桑葚样钙化并非莱迪细胞瘤的特征。
小体积莱迪细胞瘤需与莱迪细胞增生鉴别。莱迪细胞增生表现为小管间莱迪细胞融合性生长,可形成小结节(通常小于 0.5cm),在睾丸切除标本中偶然发现。由于增生常继发于激素刺激,多为多灶性和双侧性。一项研究报道胰岛素样肽 3(INSL3)呈弥漫阳性可用于鉴别增生与莱迪细胞瘤,前者弥漫阳性,后者局灶阳性或阴性。
形态类似莱迪细胞瘤但发生于先天性肾上腺皮质增生(CAH)患者的双侧多灶性病变是诊断陷阱。这类病变被不同程度地称为肾上腺生殖综合征睾丸肿瘤(TTAS)和睾丸肾上腺剩余肿瘤。与莱迪细胞瘤不同,这类肿瘤起源于产生糖皮质激素的细胞,在 CAH 患者中因促肾上腺皮质激素(ACTH)水平升高而增殖。鉴于其发病机制,且部分病变经合理药物治疗后可消退,并非所有 TTAS 均为真性肿瘤性病变,手术通常不必要。18% 的 CAH 患者以 TTAS 为首发症状,凸显了将其与莱迪细胞瘤或莱迪细胞增生区分的重要性。支持 TTAS 诊断的特征包括:双侧多灶性、门部位置、丰富脂褐素色素、混杂脂肪细胞(来自肾门)、细微嗜碱性胞质颗粒、无 Reinke 结晶、雄激素受体阴性、CD56 和突触素阳性。但支持诊断的最重要依据是 CAH 病史或皮质类固醇合成异常的生化证据(如 17‑羟孕酮水平升高)。
非特指型支持细胞瘤(Sertoli cell tumor NOS)常需与莱迪细胞瘤鉴别,此时后者部分细胞的 β‑catenin 核阳性可能造成诊断困惑。如前所述,莱迪细胞瘤可见 CTNNB1 功能获得性变异,明显为亚克隆事件,表现为局灶或多灶性 β‑catenin 核阳性。这与非特指型支持细胞瘤的弥漫表达模式形成对比,后者提示克隆性分子事件。
前瞻性识别具有恶性潜能的莱迪细胞瘤亚群(约 10%)对确定哪些患者需要随访和 / 或考虑进一步治疗、哪些术后可视为治愈至关重要。既往多项研究提出多参数临床病理危险因素评估,包括年龄、淋巴管血管侵犯、坏死、肿瘤大小、睾丸外侵犯、细胞异型性、核分裂活性和增殖指数。近期一项评分系统(莱迪细胞瘤量化评分,LeSS)在小样本研究人群中可可靠区分良恶性肿瘤,需进一步外部验证。迄今为止提出的多参数评估最显著的问题可能是各因素的预测价值未加权。但就恶性风险预测而言,肿瘤坏死或大血管侵犯等表现的意义可能大于肿瘤体积大或核异型性。近年来,一些预后生物标志物被提出。其中之一是 FH 功能缺失性变异导致的延胡索酸水合酶表达缺失,这类变异在具有侵袭性组织学特征和 / 或生物学行为的肿瘤中富集,可为体细胞或胚系事件(遗传性平滑肌瘤病肾细胞癌综合征)。MDM2 扩增在临床恶性莱迪细胞瘤中频发,可通过免疫组化间接检测或荧光原位杂交直接检测。未来可能用于评估恶性风险的其他生物标志物包括 TERT 融合、microRNA‑196b‑5p 上调和广泛非整倍体。目前,建议对所有具有侵袭性组织学特征的莱迪细胞瘤进行 FH 和 MDM2 免疫组化检测,MDM2 过表达和 FH 缺失可能提示恶性风险增加。
非特指型支持细胞瘤
非特指型支持细胞瘤是第二常见的 TSCSTs,占所有睾丸肿瘤的比例小于 1%,中位发病年龄 39 岁,也可发生于青春期前儿童和老年患者。肿瘤通常为单侧小肿瘤,表现为可触及肿块或阴囊模糊症状,部分在因其他疾病进行泌尿外科检查时偶然发现。性激素过量产生相关表现罕见。
非特指型支持细胞瘤的发病机制与 CTNNB1 变异密切相关。具体而言,具有典型形态的原发性支持细胞瘤绝大多数(超过 90%)存在该基因 3 号外显子热点区功能获得性变异。转移性肿瘤中的发生率可能较低(约 50%),但数据可能因既往研究纳入融合驱动型肿瘤而受到干扰。这类肿瘤目前被视为独立实体:炎症性巢状睾丸性索肿瘤。尽管非特指型支持细胞瘤多为散发性,部分病例见于家族性腺瘤性息肉病患者或存在致病性 APC 胚系变异的患者。其中一例患者为双侧肿瘤,另一例为同侧睾丸多灶性病变。
大体上,非特指型支持细胞瘤通常为体积小(小于 5cm)、灰白至黄色、边界清晰的结节,多达三分之一的病例可含囊腔成分。形态学上(图2),肿瘤常呈现多种生长方式,多数至少局灶可见提示性索分化的结构(如小管、条索、小梁)。其他描述的生长方式包括大囊、微囊、巢状、漩涡状、实性、网状和假乳头状。肿瘤细胞胞质中等量(少于莱迪细胞瘤),从嗜酸性到透明空泡状不等。细胞核规则,圆形至卵圆形,核仁小于莱迪细胞瘤。与后者的另一区别是间质更显著,常呈致密黏液样或胶原性,不同程度玻璃样变,与经典莱迪细胞瘤的表现相反。病变体积中大量比例为厚壁玻璃样变间质的肿瘤在文献中被称为硬化性支持细胞瘤。伴印戒细胞样外观的单空泡细胞非常常见,偶见脂质化。与腺癌的印戒细胞不同,非特指型支持细胞瘤的印戒细胞不含黏液。在 2022 版世界卫生组织分类中,纯印戒细胞样形态的性索肿瘤被视为独立实体(印戒细胞间质肿瘤)。但这一观点存在争议。
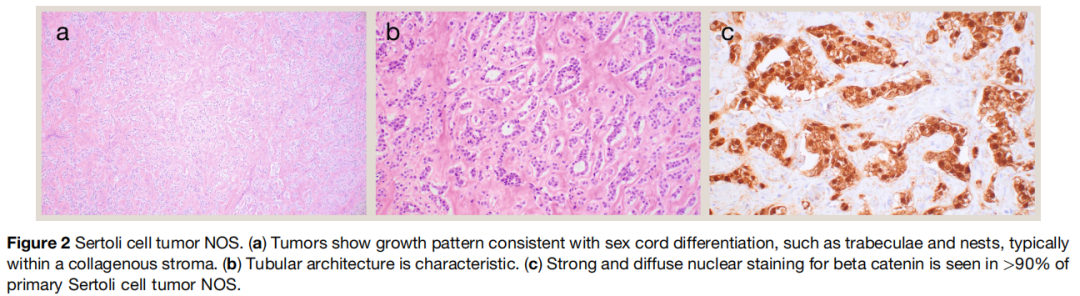
图2
非特指型支持细胞瘤常呈 SF‑1 阳性(约 80%),性索标志物(如 WT1、SOX9)和 FOXL2 阳性率约 40%–60%(灵敏度最高:SOX9,约 60%)。抑制素和钙视网膜蛋白在相当比例的病例中阳性(各约 40%)。重要的是,多数肿瘤(总体约 70%,具有典型形态的原发性肿瘤超过 90%)呈 β‑catenin 强弥漫核阳性,与 CTNNB1 变异在非特指型支持细胞瘤中的驱动作用一致。纯印戒细胞样形态的肿瘤也一致显示 β‑catenin 核阳性及潜在CTNNB1 变异,提示其可能属于非特指型支持细胞瘤谱系。
以往约 10% 的支持细胞瘤表现出临床恶性行为,但该数据早于融合驱动型肿瘤的识别,后者将在下文作为独立实体讨论。回顾性研究已确定恶性危险因素,但大多缺乏验证。提出的预测参数包括患者年龄、肿瘤大小、坏死、细胞异型性、淋巴管血管侵犯和核分裂活性。从基因组角度看,排除携带 EWSR1::ATF1 或 EWSR1 重排的病例后,恶性支持细胞瘤呈现多重非整倍体,以及致病性扩增和纯合缺失事件。
炎症性巢状睾丸性索肿瘤(INTSCT)
2002 年,Henley JD 等报道了一组临床恶性的性索肿瘤,其形态与精原细胞瘤重叠,常被误诊为精原细胞瘤。这类肿瘤发病年龄范围广,当时被归类为恶性非特指型支持细胞瘤,可能是因为其结构特征提示性索分化。最常见的转移部位包括腹膜后淋巴结和肺,此后也有骨等其他部位转移的报道。肿瘤呈实性,以巢状结构为主,纤维胶原间质丰富,混合性炎症浸润程度不等,含不同比例的淋巴细胞、浆细胞、嗜酸性粒细胞和中性粒细胞(图3)。炎症性巢状睾丸性索肿瘤核分裂活性通常较低(每 10 个高倍视野小于 5 个),但多数病例可见坏死。部分病例可见血管侵犯和睾丸外延伸等特征。肿瘤细胞中等大小,胞质中等量,淡嗜酸性至透明空泡状,细胞核略不规则,核仁小。其免疫表型具有一定特征性,除 SF‑1、抑制素等一般性索间质标志物外,还表达 CD30 和 EMA。最初按精原细胞瘤诊断和治疗的病例对顺铂为基础的化疗反应不佳,在意料之中。近期分子研究显示这类肿瘤存在频发 EWSR1::ATF1 或 EWSR1 重排。鉴于其独特的临床病理和分子特征,这类肿瘤应被视为独立实体。支持将其与非特指型支持细胞瘤区分的另一论据是,炎症性巢状睾丸性索肿瘤均呈侵袭性,而多数支持细胞瘤临床良性。从实用角度出发,研究者认为根据上述典型形态和免疫表型特征(CD30 和 EMA 阳性)可可靠诊断炎症性巢状睾丸性索肿瘤,分子检测可作为有用的辅助手段,用于确诊或排除形态不典型的病例。
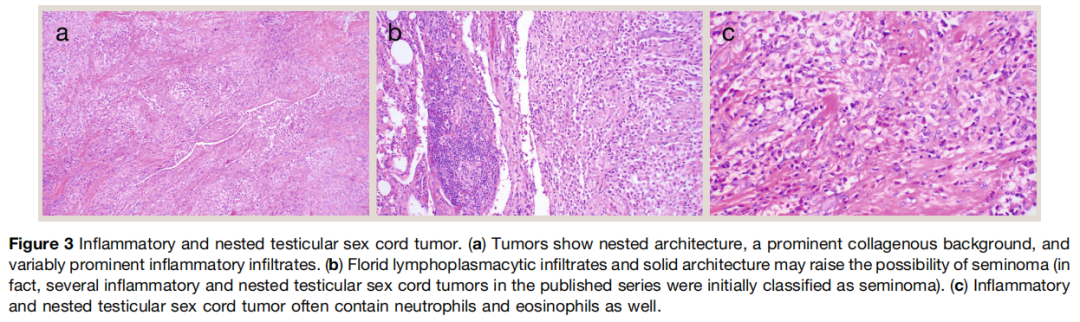
图3
大细胞钙化性支持细胞瘤
大细胞钙化性支持细胞瘤发病年龄范围广,更常见于年轻成人。约 20%–40% 的病例与 Carney 综合征相关,最常见由致病性 PRKAR1A 胚系变异导致。与 Carney 综合征相关的肿瘤通常为多灶性和双侧性,发生于儿童或青少年,临床病程惰性。偶见于神经纤维瘤病 1 型和黑斑息肉综合征患者,尽管尚不确定后者是否与小管内大细胞玻璃样变支持细胞瘤不同。综合征相关肿瘤患儿常表现为同性性早熟,而散发病例多表现为睾丸肿块,偶见男性乳房发育。约 10% 的大细胞钙化性支持细胞瘤发生转移,临床病程侵袭性。
大细胞钙化性支持细胞瘤的发病机制与蛋白激酶 A 复合物调节亚基 1A(由 PRKAR1A 编码)失活密切相关。多项病例研究和病例系列显示,无论是否与 Carney 综合征相关,大多数(超过 90%)肿瘤存在导致 PRKAR1A 失活的致病性变异。许多病例可证实该基因双等位基因失活。恶性肿瘤常出现额外基因组变异,包括其他致病性突变、非整倍体和抑癌基因纯合缺失。
大体上,典型的大细胞钙化性支持细胞瘤为边界清晰的实性结节,因存在钙化切面呈砂砾感,组织学处理前可能需要脱钙。如前所述,多灶性和 / 或双侧肿瘤应考虑 Carney 综合征相关肿瘤。镜下(图4),肿瘤可呈现多种生长方式,包括实性片块、巢状、小梁、条索和小管样结构。常可见淋巴细胞聚集,尤其在肿瘤周边。间质特征性呈黏液样,含中性粒细胞浸润和散在层状桑葚样钙化。肿瘤细胞大,多边形,胞质丰富明亮嗜酸性,细胞核圆形规则,可见核仁,与肿瘤性莱迪细胞高度相似。病变常可见小管内成分。部分肿瘤间质为纤维胶原性,钙化稀疏或无。此类情况下,大细胞钙化性支持细胞瘤与莱迪细胞瘤的鉴别困难。此时,提示性索分化的生长方式、中性粒细胞浸润和 / 或小管内成分应考虑前者。
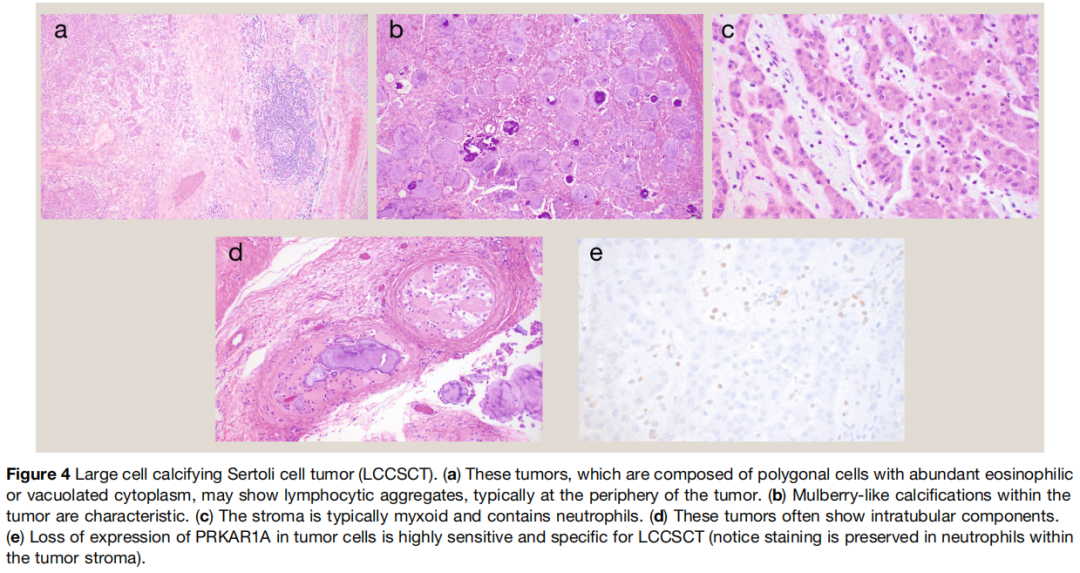
图4
抑制素和 SF‑1 是该肿瘤的灵敏标志物(两者均超过 80%),而 SOX9 和钙视网膜蛋白阳性率较低(约 50%)。WT1 仅见于少数病例(小于 20%),迄今为止检测的少量病例 FOXL2 均为阴性。总体而言,大细胞钙化性支持细胞瘤最理想的标志物是 PRKAR1A。具体而言,PRKAR1A 表达缺失区分大细胞钙化性支持细胞瘤与其最常见鉴别诊断的灵敏度为 93%,特异度为 97%。
如前所述,总体约 10% 的病例表现出恶性行为。诊断年龄是重要危险因素,大多数恶性肿瘤为散发性,发生于年龄大于 25 岁的男性。最初由 Kratzer 等描述、近期经 Al‑Obaidy 等验证的与侵袭性行为相关的组织学因素包括:肿瘤大小大于 4cm、睾丸外侵犯、坏死、细胞异型性、淋巴管血管侵犯、每 10 个高倍视野超过 3 个核分裂。
小管内大细胞玻璃样变支持细胞瘤
小管内大细胞玻璃样变支持细胞瘤始终与黑斑息肉综合征相关,最常见由 STK11 胚系突变导致。其与卵巢环状小管性索肿瘤具有相似性。这类肿瘤通常诊断于儿童或青少年,因雌激素分泌导致骨龄超前和 / 或男性乳房发育;罕见情况下可诱导同性性早熟。肿瘤为多灶性、双侧性,不形成临床明显肿块。多数病变仅在镜下可见,表现为多个小管内增殖的大多边形细胞,胞质丰富嗜酸性,小管周围基底膜增厚,可见细胞外基底膜小球,可发生钙化(图5)。受累小管缺乏成熟精子发生。尽管该实体有其名称,现已认识到部分黑斑息肉综合征相关性索肿瘤可出现浸润性成分。研究者认为其中至少部分既往被报道为大细胞钙化性支持细胞瘤,尽管其可能属于小管内大细胞玻璃样变支持细胞瘤谱系。因此,大细胞玻璃样变支持细胞瘤的命名可能更准确。
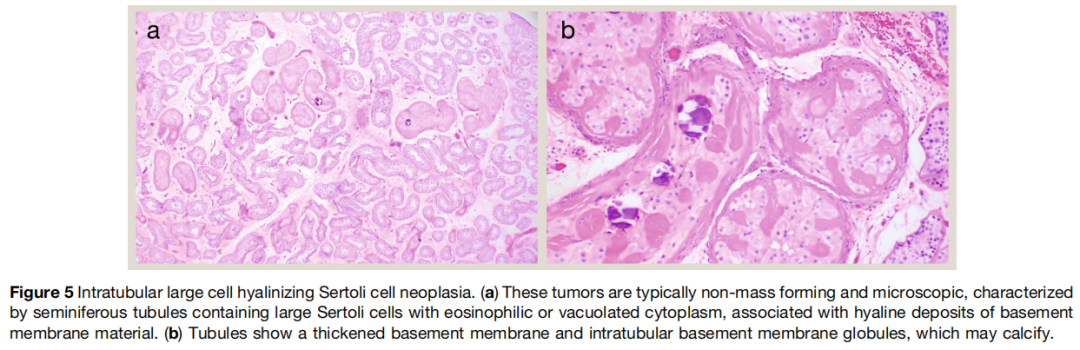
图5
小管内大细胞玻璃样变支持细胞瘤的常见鉴别诊断包括性腺母细胞瘤和支持细胞结节,两者均局限于小管 / 性索,含基底膜小球或钙化。与小管内大细胞玻璃样变支持细胞瘤不同,支持细胞结节由未成熟支持细胞构成,胞质少、淡嗜酸性至透明,可包裹精原细胞。性腺母细胞瘤含未成熟性索细胞和不同程度异型的精原细胞或明显的无性细胞瘤。正如预期,鉴于其恒定与黑斑息肉综合征相关,约 60% 的小管内大细胞玻璃样变支持细胞瘤可见 LKB1(STK11)表达缺失。
成人型颗粒细胞瘤
睾丸成人型颗粒细胞瘤(成人型粒层细胞瘤)是形态学上与卵巢同名肿瘤相似的性索肿瘤。与卵巢成人型颗粒细胞瘤(女性最常见的性索间质肿瘤)不同,睾丸成人型颗粒细胞瘤非常罕见。平均诊断年龄 40 岁,常表现为肿块,偶见男性乳房发育。与其他 TSCSTs 类似,仅较小比例(约 10%)临床恶性。
成人型颗粒细胞瘤的发病机制尚不清楚。驱动绝大多数(超过 90%)卵巢成人型颗粒细胞瘤的 FOXL2 p.C134W 热点突变在睾丸同名肿瘤中极为罕见。事实上,睾丸成人型颗粒细胞瘤的基因组特征高度异质,提示根据形态学归类的此类肿瘤可能并非单一实体。以梭形细胞成分为主的肿瘤显示出多条染色体获得模式,与其他富含梭形细胞的睾丸性索间质肿瘤相似。少数病例中发现 CTNNB1 和 NRAS 功能获得性变异。迄今为止,仅两例经分子分析的真性睾丸成人型颗粒细胞瘤存在 FOXL2 p.C134W 突变。这类肿瘤最常见的频发性事件是 22q 拷贝数丢失,这本身不太可能代表致癌事件。
大体上,肿瘤为实性或囊实性,分叶状,黄褐至白色,边界清晰。镜下,肿瘤呈现相对广泛的生长方式谱,多数反映其性索表型,包括巢状、岛状、束状、小梁 / 条索状、脑回状、微滤泡状(伴或不伴 Call‑Exner 小体)、大滤泡状、栅栏状和假乳头状。肿瘤细胞通常为小至中等大小,圆形或梭形,细胞核不规则,染色质开放,核仁小,可见核沟(常见但非特异性特征)。细胞胞质相对较少,导致核质比高,低倍镜下肿瘤呈蓝色外观。相当比例的肿瘤每 10 个高倍视野可见≥5 个核分裂,尚不确定这是否具有预后意义。睾丸外组织浸润性生长、坏死、淋巴管血管侵犯、肿瘤体积大(如大于 4cm)和显著核多形性等表现提示恶性风险。尽管部分形态学特征与卵巢肿瘤相似,睾丸同名肿瘤存在多处差异,包括 FOXL2 热点突变频率极低和激素过量相关表现。研究者推测,该分类中当前的基因组和临床病理特征可能是由于对原本无法分类的肿瘤宽松使用该诊断类别所致。
免疫组化无特异性,成人型颗粒细胞瘤表达上述一般性索间质标志物。具体而言,SF‑1、抑制素、FOXL2 和 WT1 恒定阳性,而 SOX9 常阴性。重要的是,大多数成人型颗粒细胞瘤 β‑catenin 核阴性,这一特征在需要与非特指型支持细胞瘤鉴别时可能有用。
从实用角度出发,富含梭形细胞、呈束状结构的成人型颗粒细胞瘤难以与纤维瘤 / 卵泡膜细胞瘤和肌样性腺间质肿瘤鉴别。此时网状组织化学染色可能有用,纤维瘤中网状纤维围绕单个细胞,成人型颗粒细胞瘤中围绕细胞群。成人型颗粒细胞瘤以性索成分为主时最主要的鉴别诊断是非特指型支持细胞瘤。β‑catenin 免疫组化有助于区分这两类肿瘤,鉴于罕见睾丸成人型颗粒细胞瘤可出现 CTNNB1 功能获得性突变。
幼年型粒层细胞瘤
睾丸幼年型粒层细胞瘤(青少年型粒层细胞瘤)均发生于儿童患者,超过 90% 在出生后第一年内诊断。与卵巢幼年型粒层细胞瘤不同,睾丸幼年型粒层细胞瘤大多无激素活性,且均为良性。表现为临床明显肿块,可为先天性(出生前或出生时发现)。
睾丸幼年型粒层细胞瘤的发病机制尚不清楚。部分病例见于性腺发育不良的情况。卵巢肿瘤中所见的变异,包括 AKT1 的 PH 结构域内部串联重复和 GNAS 热点突变,在睾丸肿瘤中未见报道。相反,约 60% 的病例显示 10 号染色体单体。
大体上,睾丸幼年型粒层细胞瘤边界清晰,多囊性或囊实性,纤维分隔将囊腔分开。组织学上(图6),典型肿瘤含大小不等的滤泡,内含嗜碱性或嗜酸性稀薄分泌物。滤泡衬覆小卵圆形至星芒状细胞,细胞核小,胞质少嗜酸性。囊腔 / 滤泡与实性区交替分布,实性区含梭形细胞和与滤泡衬覆细胞相似的圆形至星芒状细胞,常伴细胞间嗜碱性或嗜酸性分泌物积聚。间质从纤维胶原性到黏液样不等。核分裂可丰富,但无预后意义。免疫组化无特异性,睾丸幼年型粒层细胞瘤表达前述一般性索间质标志物。
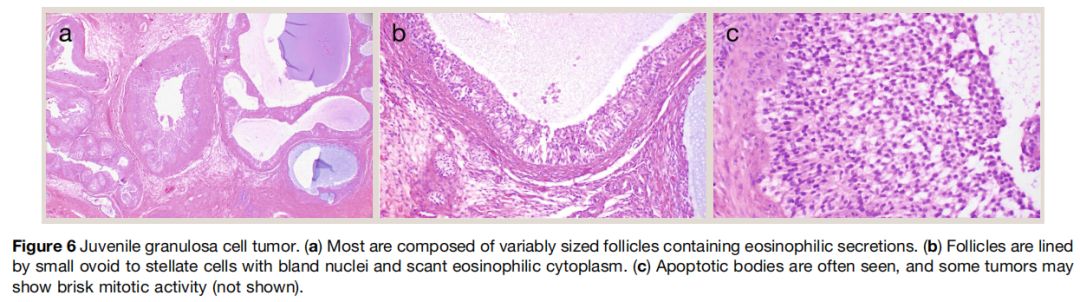
图6
鉴于其特征性临床表现,睾丸幼年型粒层细胞瘤的鉴别诊断有限。发生于年幼儿童的多囊性形态需考虑青春期前型卵黄囊瘤,通过有限的免疫组化 panel(如 SF‑1、抑制素和 SALL4)可轻松鉴别。其他需考虑的包括婴儿型支持细胞瘤,尽管该区分可能存在一定主观性。
鉴于卵巢和睾丸幼年型粒层细胞瘤之间重要的临床病理和分子差异,有建议将后者命名为睾丸良性幼年型粒层细胞瘤。或者,报告中可加入解释性说明,指出这类肿瘤均呈惰性,凸显其与卵巢粒层细胞瘤的重要区别,可能避免不必要的积极临床处理。
纯梭形细胞形态的间质肿瘤
WHO 分类认可的纯梭形细胞形态性腺间质肿瘤包括两个实体:肌样性腺间质肿瘤和纤维瘤 / 卵泡膜细胞瘤。两者均发生于青春期后患者,表现为肿块,无性激素产生相关表现。这类肿瘤均为良性;因此,其区分更多具有学术意义而非临床意义。
其发病机制大多未知。少数研究采用基因组 DNA 测序和荧光原位杂交显示,这类间质肿瘤呈现多条染色体获得模式,符合倍性改变。未发现复发性突变和基因融合。
大体上,肿瘤边界清晰,无包膜,实性结节,切面漩涡状、灰白至褐色。肌样性腺间质肿瘤起源于原始小管周围肌样细胞,定义性共表达 S100 和 SMA(图7)。纤维瘤可发生于实质内(类似卵巢纤维瘤)或白膜中心(即白膜纤维瘤)。两类肿瘤均由形态温和的梭形细胞呈束状、漩涡状或鱼骨样排列构成。常包裹生精小管,部分肿瘤每 10 个高倍视野可见多达 9‑10 个核分裂,但这似乎与侵袭性行为无关。纤维瘤的胶原性间质通常比肌样性腺间质肿瘤更显著,包括胶原斑块的存在。
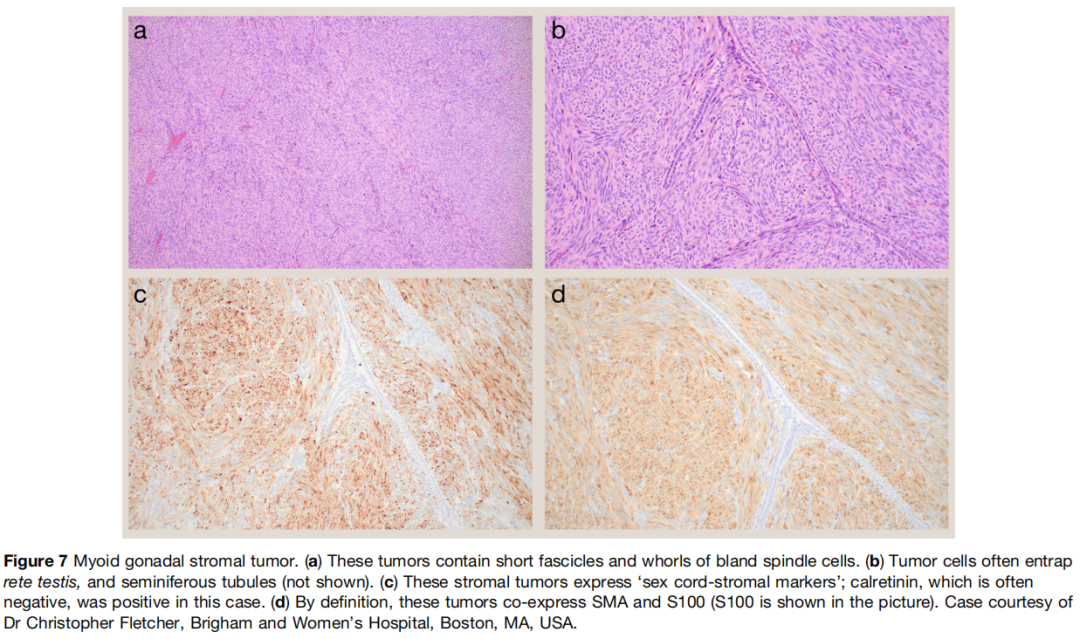
图7
肌样性腺间质肿瘤和纤维瘤 / 卵泡膜细胞瘤均表达性索间质标志物;值得注意的是,肌样性腺间质肿瘤一致报告 SOX9 阴性,而大多数纤维瘤阳性。如前所述,S100 与 SMA 共表达是肌样性腺间质肿瘤的必要条件。但尚无关于做出该诊断所需 S100 表达程度的标准;鉴于 S100 是多种性索间质肿瘤类型常表达的非特异性标志物,这可能造成诊断困难。鉴于其临床病理相似性,当无法明确区分肌样性腺间质肿瘤和纤维瘤 / 卵泡膜细胞瘤时,诊断梭形细胞性腺间质肿瘤是恰当的。同时加入说明,指出迄今为止所有报道病例均呈惰性生物学行为,可能也是合适的。
混合型性索-间质肿瘤和性索-间质肿瘤,非特殊类型
混合型性索-间质肿瘤是指包含性索和间质成分混合的肿瘤(图8)。部分文献中因其性索结构(小管、条索、小梁)与肿瘤性梭形细胞间质成分混合而被称为支持‑间质细胞瘤。极其罕见的睾丸支持‑莱迪细胞瘤也可归入该类别,鉴于莱迪细胞为间质起源。重要的是,应将包裹非肿瘤性莱迪细胞的性索肿瘤(相对常见)与莱迪细胞成分也为肿瘤性的真性混合性肿瘤(睾丸中极其罕见)区分开。据研究者所知,仅一例睾丸支持‑莱迪细胞瘤报道携带 DICER1 变异,该特征是卵巢支持‑莱迪细胞瘤主要亚群的特征。
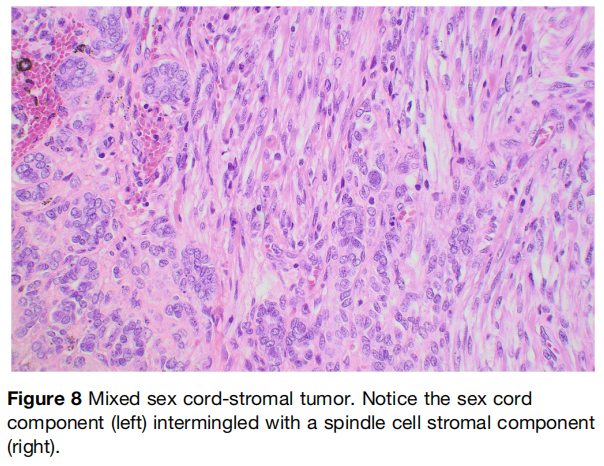
图8
TSCSTs NOS是指无法归入前述任何亚型的纯性腺或性索肿瘤。根据定义,该类别具有异质性,包括临床良性和恶性肿瘤。分子研究或免疫组化可能有助于提示形态学上不明显的肿瘤类型。
结 论
TSCSTs 的诊断、分类和预后评估具有挑战性,需要经验。准确报告这类肿瘤对临床管理具有重要意义,包括遗传性肿瘤易感综合征的评估。近期,由泌尿病理学会(GUPS)和国际泌尿病理学会(ISUP)认可的一组泌尿病理医师发布了 TSCSTs 分类和检查指南。尽管本文提及了最相关的建议,研究者建议读者参考该近期出版物获取更多细节。结合临床、组织病理和分子特征(如可获得)的整合方法可最大限度提高准确分类的可能性。未来,更大规模的中心会诊研究将是确定 TSCSTs 更准确风险预测因子(包括生物标志物)的关键。
参考文献:
Lobo J, Acosta AM, An update on testicular sex cord stromal tumours: recent advances and diagnostic pearls, Diagnostic Histopathology, https://doi.org/10.1016/j.mpdhp.2026.02.002