首页 > 健康资讯/ 正文
深度解析医学证据,lxfs.net为你支撑决策
自人类基因组计划完成以来,全球多项大规模全基因组关联(GWAS)研究也相继展开,目前已鉴定出大量与复杂性状和疾病相关的遗传位点,极大促进了人类遗传学研究。然而,该领域长期存在严重的族群样本失衡问题,现有研究大多以欧洲血统(EUR)人群为主,现有数据显示仅约12%研究受试者为非欧洲血统,这不仅限制了对通用和族群特异性遗传位点的发现,也削弱了多基因风险评分等研究成果在不同祖先群体间的可迁移性和临床预测效能。为打破欧洲人群数据垄断,近年来中国、日本、韩国等陆续搭建了本土生物样本库,提供了宝贵的亚群特异性遗传信息,但大多数队列规模偏小,且个体级数据与完整统计数据获取受限,东亚人群跨队列、跨性状的系统性遗传研究始终难以深入推进。
近日,中国台湾、韩国、日本等研究团队合作开展了迄今规模最大的东亚(EAS)人群表型组荟萃分析,共纳入超103万名受试者,涵盖127种复杂性状。研究识别出8,010个全新遗传关联位点,发现东亚各亚群间存在广泛遗传共享,同时也检测到不同队列间的遗传异质性。跨种族分析显示,东亚与欧洲人群之间存在中等程度的遗传相关性,表明疾病风险存在共享与族群特异性遗传组分;多效性分析揭示人类白细胞抗原(HLA)区域内存在显著遗传信号。通过对全基因组关联矩阵进行分解,研究进一步揭示了结构化的跨表型架构,呈现出一个主要由代谢、生化和人体测量性状驱动的共享多基因骨架,以及两个富集于免疫相关过程的潜在遗传组分。总之,该研究系统描绘了东亚人群复杂性状的共享与特异性遗传架构。相关内容发表于预印本平台bioRxiv。

该研究围绕东亚人群11大类共127项性状展开分析,整合了来自日本、韩国、中国大陆及台湾地区7个大型队列,涵盖1,035,254名参与者。其中,约75%参与者来自汇总统计数据,约25%来自个体级基因分型数据。东亚人群定量性状中,收缩压样本量最大,分类性状中高血压/降压药使用样本量最大;同时纳入欧洲人群数据作为对照,身高、肝硬化分别为欧洲人群样本量最高的两类性状。东亚和欧洲数据集总计纳入超过200万名个体,为大规模东亚人群及跨种族遗传结构鉴定提供支持。
研究团队先对各东亚亚人群分组开展GWAS,不同地区人群检出数量各异的显著遗传变异,经连锁不平衡过滤后获得大量独立遗传信号,关联结果多集中于A至C类数量性状,身高相关位点在多个东亚亚人群中检出最多。随后,研究团队设置了两种荟萃分析方案(CJKT和JKT),共得到596,905个全基因组显著关联结果,包含14,976个独立遗传信号、8,010个未被报道的全新遗传位点,新位点主要富集于A-C类(代谢、生化、体型相关性状)多基因数量性状,在F类分类性状(免疫相关)中也检出部分遗传信号。
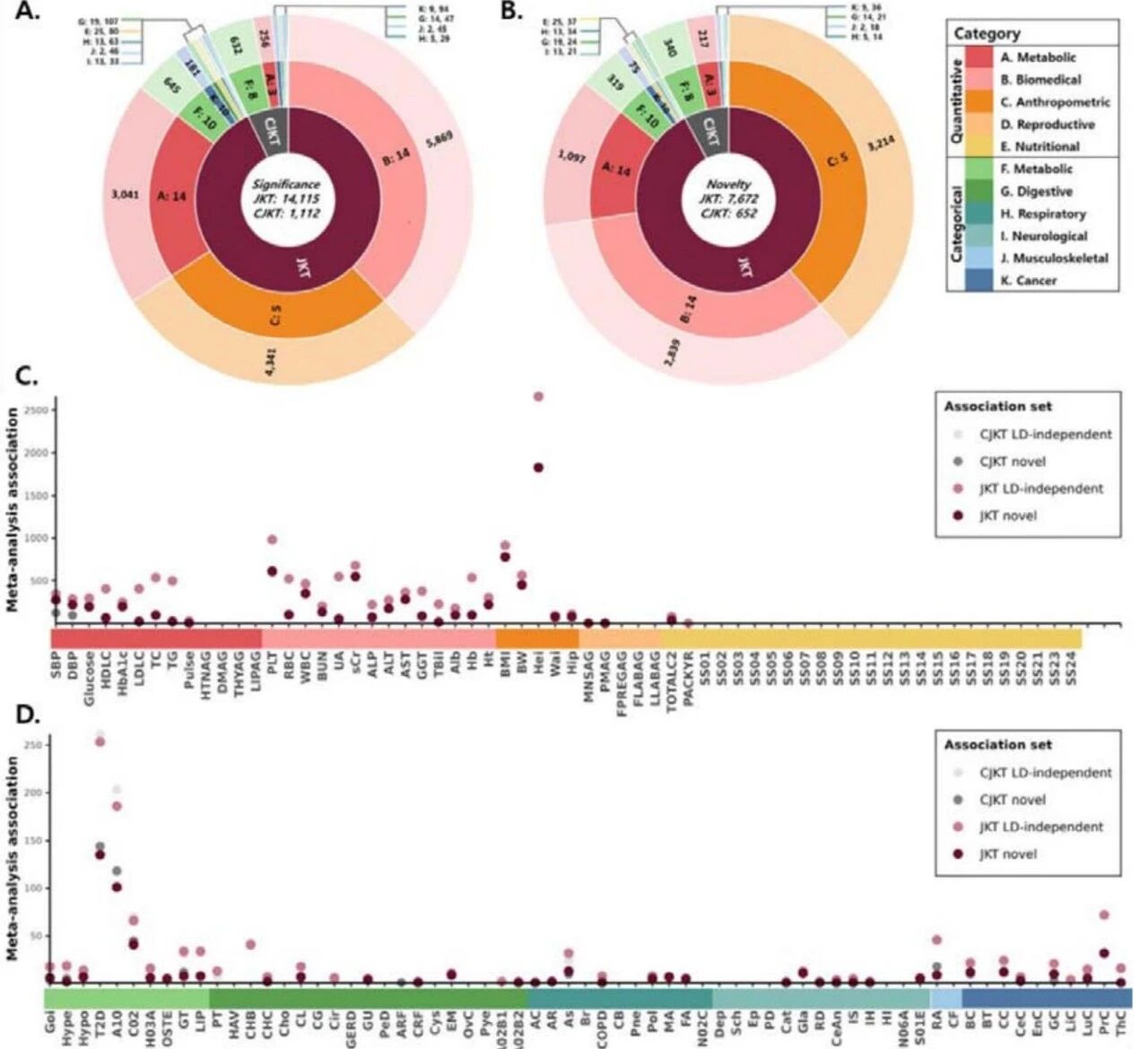
图1. EAS荟萃分析中的全基因组显著及新发现的关联
接下来,研究团队从遗传力、遗传相关性和基因水平系统解析了东亚人群复杂性状的遗传结构。在遗传力方面,东亚不同队列间定量性状的SNP遗传力估计值总体一致,而分类性状估计值普遍偏低;部分小样本队列易出现遗传力估算不稳定的情况。在东亚亚人群及全东亚人群荟萃分析中,SNP遗传力估计值整体相近,但略低于单一大型队列;欧洲人群遗传力整体高于东亚,身高为两类人群遗传力最高性状,各类分类性状遗传力均趋近于0。
在遗传相关性方面,东亚各亚人群间的定量性状遗传相关性较高,但涉及中国嘉道理生物样本库(CKB)的模式与其他亚群存在差异,可能源于地理多样性招募及分析方法不同。跨种族分析显示,东亚与欧洲人群对于76种有效性状的遗传相关系数约0.8,表明复杂性状的遗传结构在不同祖先群体间具有广泛共享性。基因水平分析(JKT队列)显示,鉴定出5,901个基因与65种性状之间的12,899个显著关联信号,93.6%集中于A-C类性状,身高相关基因位点多达2,451个,印证了该性状的多基因遗传特性。
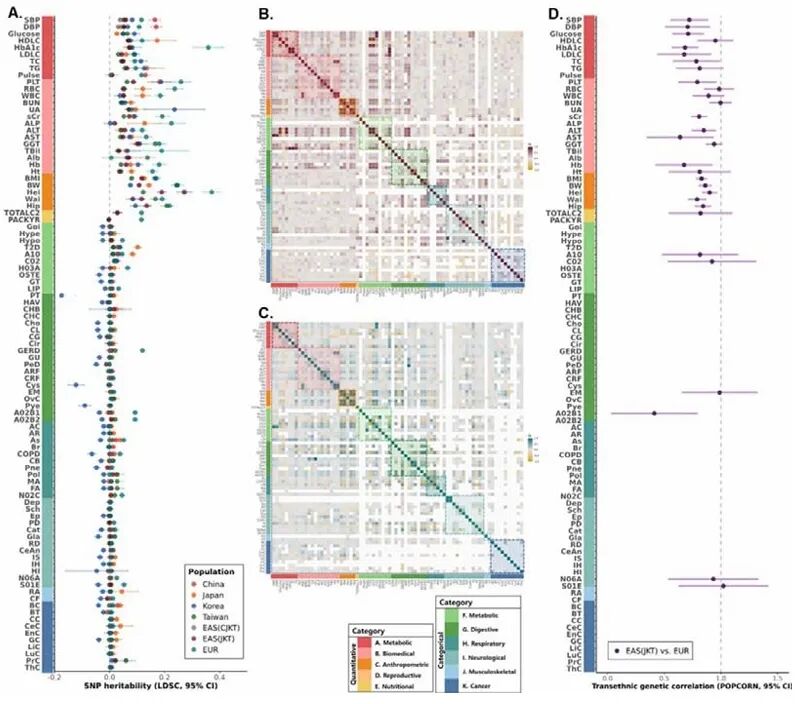
图2. EAS与EUR群体之间的遗传结构比较
研究团队从变异和基因水平对东亚人群复杂性状的多效性关联进行了系统分析。在包含主要组织相容性复合体(MHC)区域的分析中,东亚人群(JKT)荟萃分析针对76种表型共产生589,475个全基因组显著关联,涉及379,457个SNP,其中39个变异位点与超20种性状相关,被定义为多效性位点,主要分布在4个基因组区域:2号染色体GCKR/ZNF512、6号染色体HLA、9号染色体ABO以及12号染色体CUX2/ATXN2/NAA25/PTPN11区域。多数候选基因位于HLA区域,这些基因构成高度富集的蛋白质互作网络并形成5大功能簇。
基因水平分析显示,鉴定出5,901个基因和65种性状之间的12,899个显著基因-性状关联,主要富集于A -C类性状,其中31个基因可调控12个以上性状,为多效性基因,分布于8条染色体,覆盖了变异水平鉴定的所有多效性区域,两种方法存在12个共有多效性基因。该组基因同样形成显著富集的蛋白互作网络,划分出5个基因簇。
对两个水平多效性基因并集进行分析,共得到49个独特基因,其形成显著富集的蛋白互作网络,可分为8个基因簇并存在簇间连接。通路富集分析,在65种性状中鉴定出5,407条富集通路,类风湿关节炎、生化类性状分别在个体性状、性状类别中富集通路最多;富集结果主要集中在免疫相关通路,特别是与病毒感染、抗原加工和呈递以及自身免疫性疾病相关的通路,证实了HLA区域在东亚人群多种复杂性状遗传调控中的关键作用。
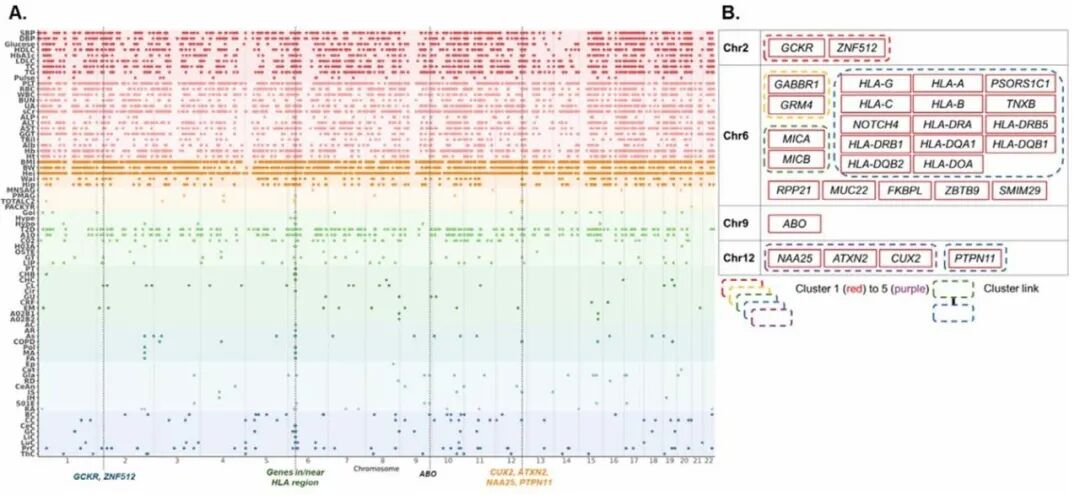
图3. EAS(JKT)荟萃分析中鉴定出的多效基因位点及其相互作用网络
研究团队基于累计方差解释率70%阈值筛选得到16个遗传潜在成分,多数成分由代谢、生化、人体测量等性状共同驱动,构成东亚人群通用多基因遗传骨架;同时存在疾病特异性遗传模块,LC10和LC15分别由青光眼和抑郁相关性状主导。CSMD1、RBFOX1等多个关键基因广泛参与多类性状调控,经通路富集可形成功能统一的基因簇。不同疾病特异性LC对应独特的生物学通路,如LC4、LC10、LC15、LC16等分别富集于免疫调控、视网膜神经发育、大脑皮层兴奋性神经传递与肿瘤易感相关通路。
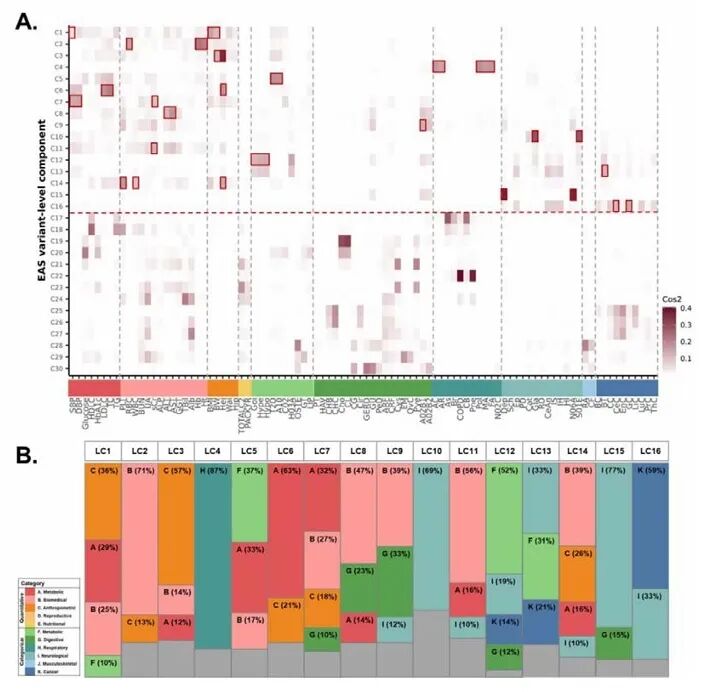
图4. 潜在成分间的特征贡献分数模式及类别层面的贡献
综上,该研究是目前规模最大的东亚人群全表型组荟萃分析,首次系统构建百万级东亚人群多性状遗传数据库,一次性挖掘8,010个全新遗传位点,弥补欧洲人群主导下基因组研究的族群失衡缺陷。研究厘清东亚内部亚人群遗传共性与异质性,明确HLA区域为免疫类性状核心多效调控区域,为全球基因组多样性研究提供关键东亚参照体系。
参考文献:
https://www.biorxiv.org/content/10.64898/2026.06.18.730290v1
小提示:本篇资讯仅在梅斯医学APP中开放阅读,请扫描二维码直接下载APP
