深度解析医学证据,DeepEvidence为你支撑决策
背景介绍
肝缺血再灌注损伤(HIRI)是肝切除术和肝移植后常见且目前难以避免的病理事件,可诱发炎症反应并损害肝功能,增加并发症风险并降低移植物的长期存活率。作为一种双相损伤,HIRI发生在肝脏血供暂时中断随后恢复的过程中,会引发活性氧过量产生、炎症反应和细胞死亡等一系列级联事件。然而,目前针对HIRI的有效预防和治疗策略仍十分有限。B细胞易位基因2(Btg2)属于BTG/TOB抗增殖基因家族,最初被认为是一种受生长抑制信号激活的即刻早期基因,其肿瘤抑制特性备受关注。近年来,Btg2已被证实通过调节ROS水平,成为维持细胞稳态的多功能介质。然而,Btg2在HIRI中的具体作用尚不明确。铁死亡是一种铁依赖性的调节性细胞死亡形式,以脂质过氧化产物和ROS的积累为特征,已被证实参与缺血再灌注损伤等多种病理过程。因此,深入探究HIRI中Btg2调控铁死亡的分子机制具有重要的理论意义和临床转化价值。
研究思路
针对上述科学问题,重庆医科大学附属第一医院吴忠均、Yu Huarong联合重庆大学Huang Zuotian、四川大学杨家印团队开展了一项系统的研究。通过RNA-seq分析,发现Btg2在HIRI后表达显著上调。利用肝细胞特异性Btg2转基因和全身性Btg2基因敲除小鼠模型,结合70% HIRI模型,证实Btg2会加重肝脏炎症和细胞凋亡。代谢组学分析显示,Btg2敲除小鼠肝脏中牛磺酸代谢受到最显著的影响。机制上,Btg2通过与含黄素单加氧酶1(Fmo1)相互作用,抑制E3连接酶Ufl1介导的Fmo1的UFMylation修饰,从而促进Fmo1发生K48连接的泛素化降解,导致Fmo1蛋白水平下降,限制牛磺酸合成,进而加剧铁死亡和细胞凋亡。通过虚拟筛选天然化合物,发现Daturataturin A(DTA)能够有效靶向抑制Btg2,从而减轻铁死亡和HIRI。相关内容以Btg2 suppresses UFMylation of Fmo1 to promote ferroptosis and exacerbate hepatic ischemia-reperfusion injury by restricting taurine synthesis为题,发表在Nature Communications!

图片解析
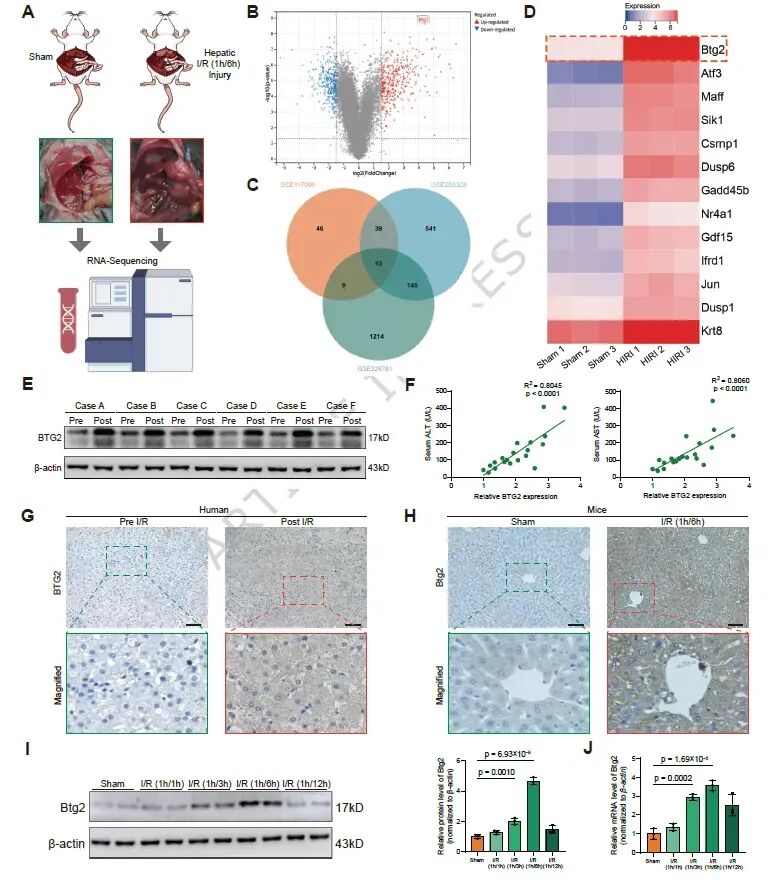
图1. HIRI中Btg2表达上调: (a) 小鼠HIRI模型及RNA-Seq样品采集示意图。(b) HIRI与Sham组肝脏差异表达基因火山图。(c) GSE293329、GSE117066和GSE228781三个数据集的共同差异表达基因Venn图。(d) 13个共同差异表达基因的热图。(e) 人非恶性肝组织HIRI前后BTG2蛋白表达。(f) BTG2表达与术后第一天血清ALT/AST的相关性分析。(g) 人肝组织HIRI前后BTG2的IHC染色。(h) 小鼠Sham和HIRI组肝脏Btg2的IHC染色。(i,j) 小鼠肝脏中Btg2蛋白和mRNA的时间依赖性表达。结果表明Btg2在HIRI后显著上调,且与肝功能损伤正相关。
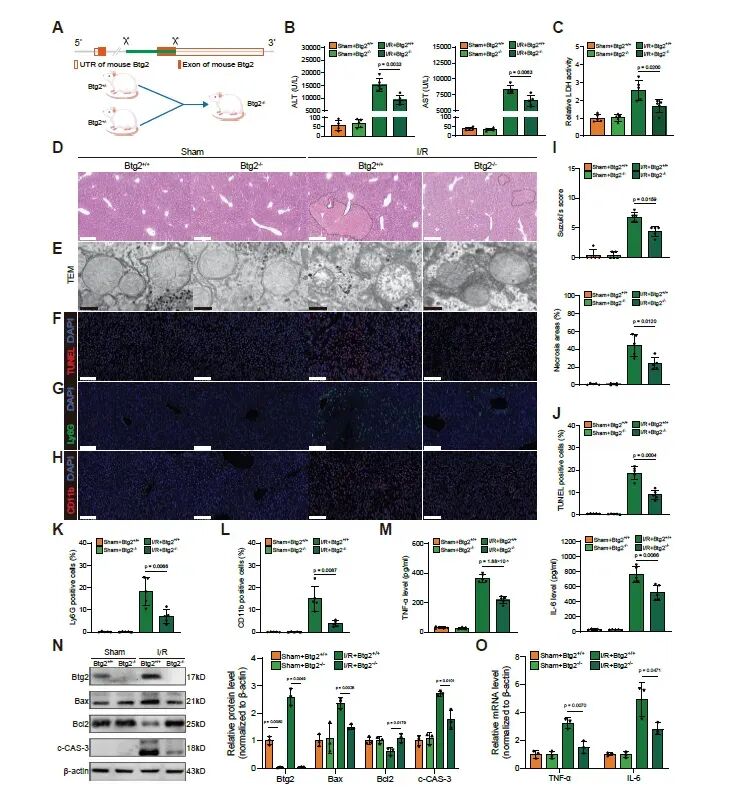
图2. Btg2敲除减轻HIRI诱导的肝细胞损伤和炎症: (a) Btg2敲除小鼠构建示意图。(b,c) 血清ALT、AST和LDH水平。(d) 肝脏H&E染色。(e) 肝细胞线粒体TEM分析。(f) TUNEL染色。(g,h) Ly6G和CD11b免疫荧光染色。(i-l) Suzuki评分、坏死面积及阳性细胞定量分析。(m) 炎症因子ELISA检测。(n) 凋亡相关蛋白Western blot。(o) 炎症因子mRNA表达。Btg2敲除显著减轻了HIRI诱导的肝损伤、炎症和细胞凋亡。
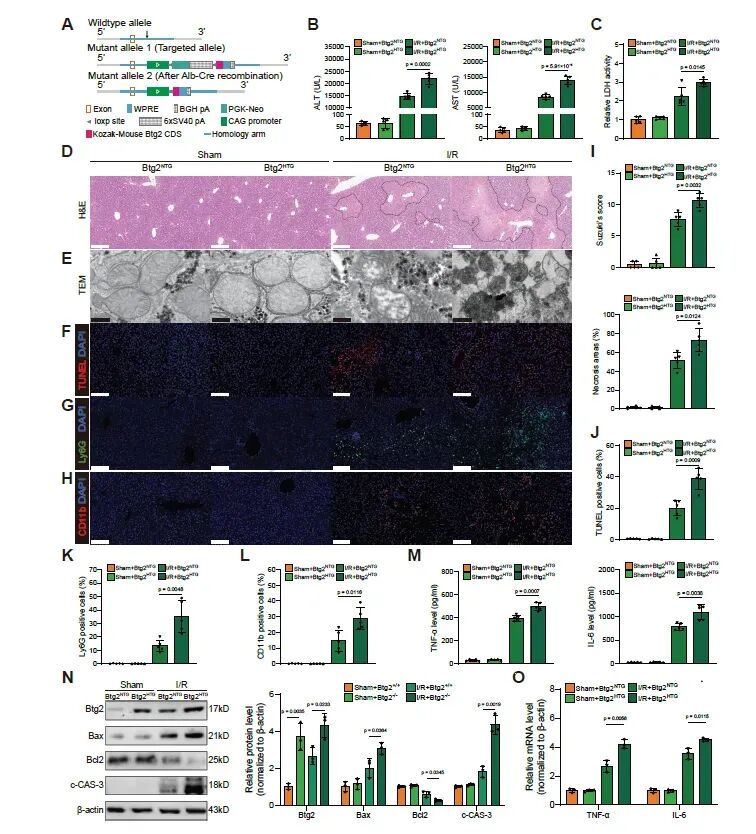
图3. 肝细胞特异性Btg2过表达加重HIRI: (a) 肝细胞特异性Btg2转基因小鼠构建示意图。(b-o) 各项指标检测结果与图2类似,但趋势相反。Btg2过表达显著加重了HIRI诱导的肝损伤、炎症、线粒体损伤和细胞凋亡。
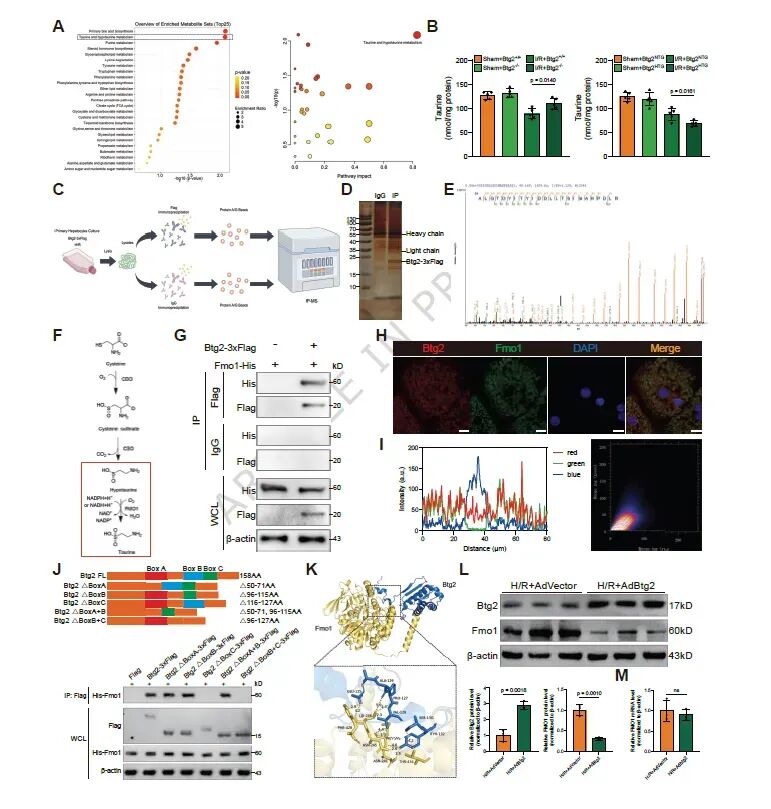
图4. Btg2与Fmo1相互作用并下调其表达: (a) 代谢组学分析显示牛磺酸和亚牛磺酸代谢是最显著影响的通路。(b) 不同小鼠肝脏中牛磺酸浓度检测。(c,d) Btg2-3xFlag免疫沉淀产物的银染和质谱鉴定。(e) Fmo1被鉴定为Btg2最可能的互作蛋白。(f) Fmo1催化亚牛磺酸转化为牛磺酸示意图。(g) Co-IP验证Btg2与Fmo1相互作用。(h,i) 免疫荧光验证Btg2与Fmo1在细胞质中共定位。(j) Btg2的Box-C结构域介导其与Fmo1的相互作用。(k) Btg2与Fmo1分子对接分析。(l,m) Btg2过表达降低Fmo1蛋白水平但不影响其mRNA水平。Btg2通过其Box-C结构域与Fmo1相互作用,负向调控Fmo1蛋白水平。
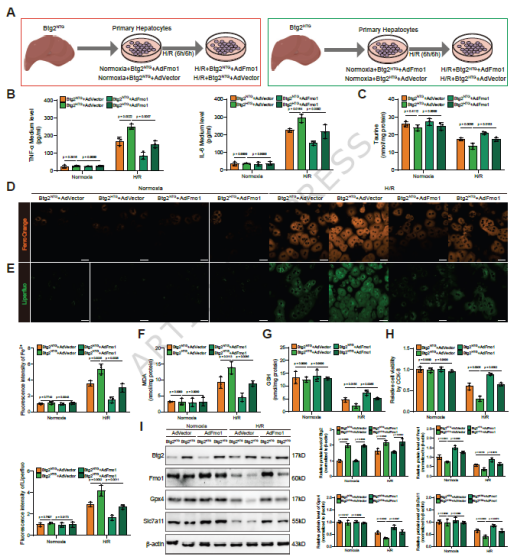
图5. Btg2通过下调Fmo1限制牛磺酸合成加剧铁死亡: (a) Btg2HTG小鼠原代肝细胞转染AdFmo1的实验设计。(b) 炎症因子ELISA检测。(c) 牛磺酸浓度检测。(d,e) Fe²⁺和LPO水平检测。(f,g) MDA和GSH浓度检测。(h) CCK-8细胞活力检测。(i) 铁死亡相关蛋白Western blot。Fmo1过表达恢复了Btg2过表达细胞中的牛磺酸合成,减轻了铁死亡和细胞损伤。
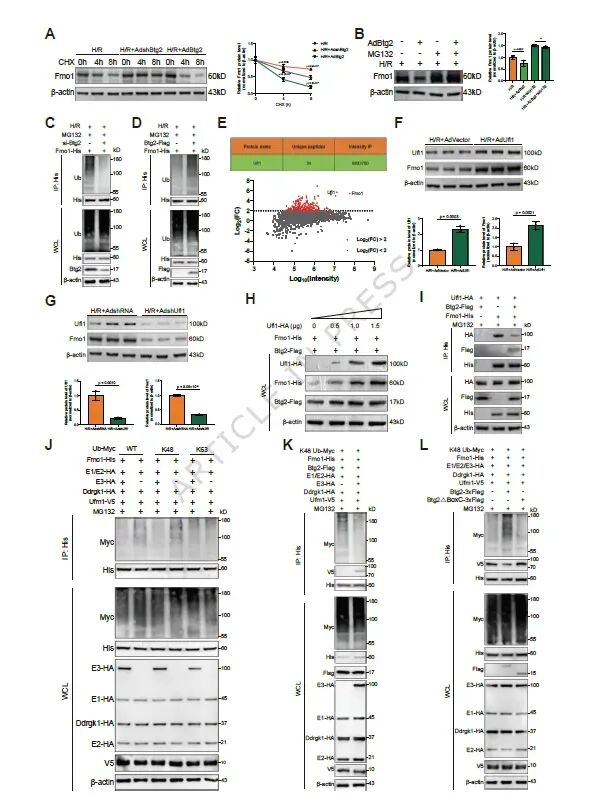
图6. Btg2抑制Fmo1的UFMylation并促进其泛素化降解: (a) CHX实验显示Btg2缩短Fmo1半衰期。(b) MG132抑制蛋白酶体后Btg2无法降解Fmo1。(c,d) Btg2促进Fmo1的K48-泛素化。(e) IP-MS鉴定可能调控Fmo1稳定性的E3连接酶Ufl1。(f,g) Ufl1过表达/敲低影响Fmo1表达。(h) Ufl1剂量依赖性稳定Fmo1。(i) Btg2抑制Ufl1与Fmo1的结合。(j) Ufl1主要抑制Fmo1的K48-泛素化。(k) Ufl1促进Fmo1的UFMylation并抑制其K48-泛素化。(l) Btg2的Box-C结构域抑制Fmo1的UFMylation并促进K48-泛素化。Btg2通过其Box-C结构域抑制Ufl1介导的Fmo1 UFMylation,从而促进Fmo1的K48-泛素化降解。
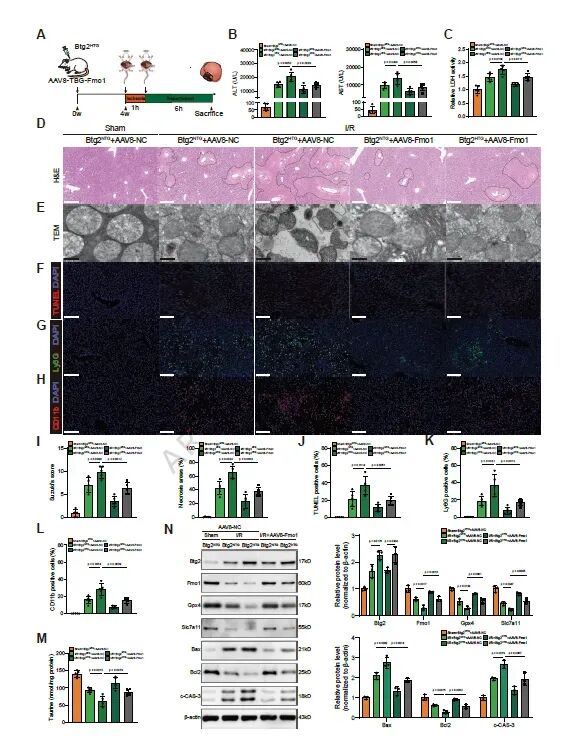
图7. 体内Fmo1过表达挽救Btg2HTG小鼠的HIRI表型: (a) AAV8-Fmo1体内实验设计。(b-d) 血清ALT、AST和LDH水平。(e) H&E染色。(f) TEM分析。(g) TUNEL染色。(h,i) Ly6G和CD11b染色。(j-l) Suzuki评分、坏死面积及阳性细胞定量。(m) 肝脏牛磺酸浓度检测。(n) 凋亡和铁死亡相关蛋白Western blot。体内Fmo1过表达显著逆转了Btg2HTG小鼠的HIRI表型。
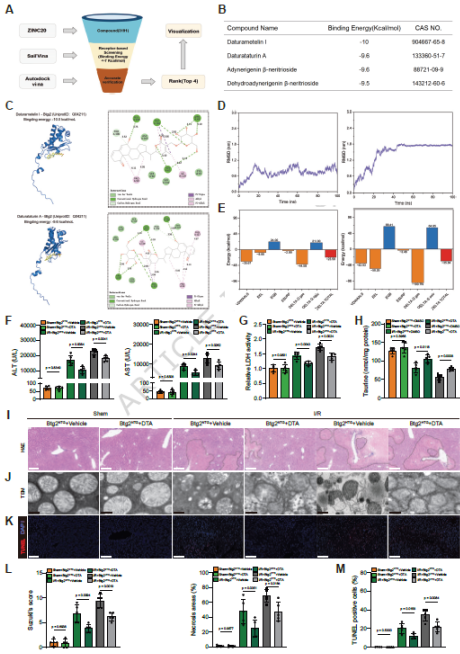
图8. DTA通过靶向Btg2减轻HIRI: (a) 天然化合物虚拟筛选示意图。(b) 与Btg2结合能<-9 kcal/mol的4种化合物。(c) Btg2与DTA/DTI的分子对接预测。(d) Btg2-配体复合物主链原子RMSD。(e) 分子动力学模拟的氨基酸能量分解。(f,g) 血清ALT、AST和LDH水平。(h) 肝脏牛磺酸浓度检测。(i) H&E染色。(j) TEM分析。(k) TUNEL染色。(l,m) Suzuki评分和坏死面积定量。DTA通过靶向抑制Btg2,恢复了牛磺酸水平,减轻了HIRI。
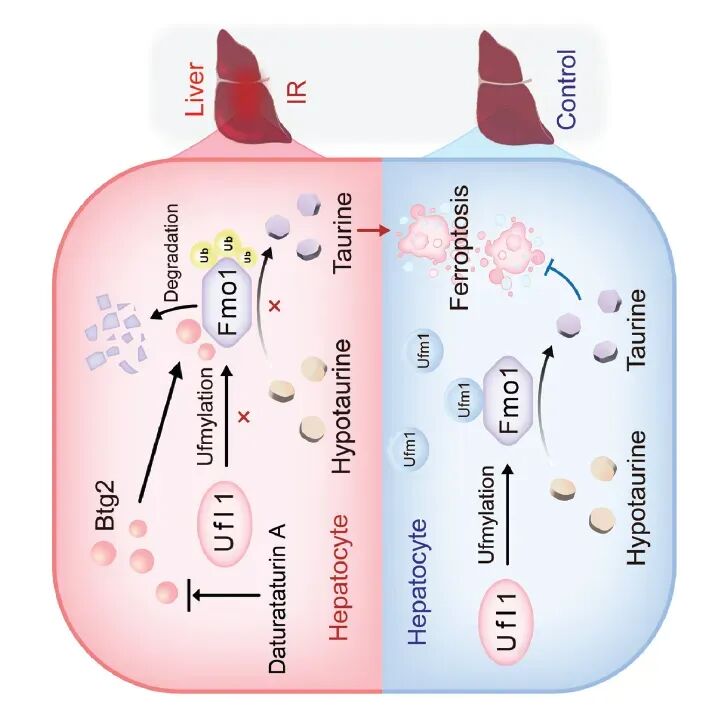
图9. 总体机制示意图: Btg2在HIRI中上调,通过其Box-C结构域与Fmo1相互作用,抑制Ufl1介导的Fmo1 UFMylation,促进Fmo1的K48-泛素化降解,导致Fmo1蛋白水平下降,限制亚牛磺酸向牛磺酸的转化,牛磺酸缺乏加剧了铁死亡和细胞凋亡,最终加重HIRI。天然化合物DTA可通过抑制Btg2来减轻HIRI。
结论
本研究表明,Btg2在HIRI中发挥着关键的促损伤作用。在HIRI条件下,Btg2表达显著上调。通过其Box-C结构域与Fmo1相互作用,Btg2抑制了E3连接酶Ufl1介导的Fmo1的UFMylation修饰,转而促进Fmo1发生K48连接的泛素化降解,导致Fmo1蛋白水平下降,进而限制了亚牛磺酸向牛磺酸的转化。牛磺酸的缺乏加剧了铁死亡和细胞凋亡,最终加重了HIRI。通过全基因敲除和肝细胞特异性转基因小鼠模型,研究团队分别在体内和体外证实了Btg2的促损伤作用。此外,通过虚拟筛选从天然化合物中鉴定出DTA能够有效靶向抑制Btg2,从而减轻铁死亡和HIRI。这些发现揭示了Btg2在HIRI中的新功能,连接了UFMylation修饰与牛磺酸代谢,表明Btg2可能是HIRI的一个有前景的治疗靶点。
原文链接:
https://www.nature.com/articles/s41467-026-72455-z