深度解析医学证据,DeepEvidence为你支撑决策
在神经退行性疾病领域,特别是以tau蛋白异常聚集为特征的疾病(如阿尔茨海默病、额颞叶痴呆、慢性创伤性脑病等),免疫系统的作用日益成为研究焦点。传统上,伴随病变产生的神经炎症常被视为加剧神经元损伤的“帮凶”。美国国家神经疾病与中风研究所Dorian B. McGavern团队在《Nature Immunology》发表的最新研究成果,系统揭示了在tau蛋白病发展中,先天免疫与适应性免疫系统之间存在着一种有序的协同防御机制。该机制的核心是一类独特的GZMK+CD8+T细胞,它们通过特异性靶向并清除功能失调的小胶质细胞,从而发挥延缓疾病进展的保护性作用。这一发现在转基因小鼠模型和人类患者脑组织中得到相互印证,为理解神经免疫互作提供了全新的理论框架。

研究首先在表达人类P301S突变tau蛋白的转基因小鼠模型中,利用高维流式细胞术全面解析了疾病进展期的脊髓免疫细胞组成。如图1a, b所示,通过Uniform Manifold Approximation and Projection降维可视化与定量分析发现,与同窝野生型对照相比,出现明显神经症状的纯合子小鼠脊髓中,免疫微环境发生了显著变化。其中,活化的CD11c+及CD11c+MHCII+小胶质细胞亚群数量显著增加,同时CD8+与CD4+T细胞也大量浸润。这种免疫细胞的募集与异常磷酸化tau蛋白在脊髓中的高负荷分布呈现出空间关联性(图1c-f),提示免疫系统正对tau病理产生主动应答。
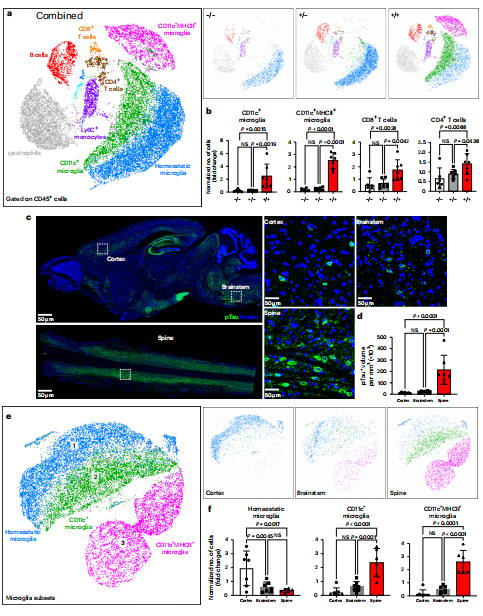
图1 Tau蛋白病变促进免疫细胞的募集和激活
为进一步阐明其分子基础,研究人员对病变脊髓组织进行了批量RNA测序。主成分分析与聚类热图(图2a, b)显示,纯合子小鼠的基因表达谱与对照组截然不同。通路富集分析(图2c, d)揭示,病变组织中抗原提呈、补体激活、白细胞介素信号及整合素信号等与免疫激活密切相关的通路被显著上调。这从转录组水平证实,tau蛋白病在中枢神经系统内引发了一场协调的神经炎症反应,并暗示小胶质细胞可能正向抗原提呈细胞分化。
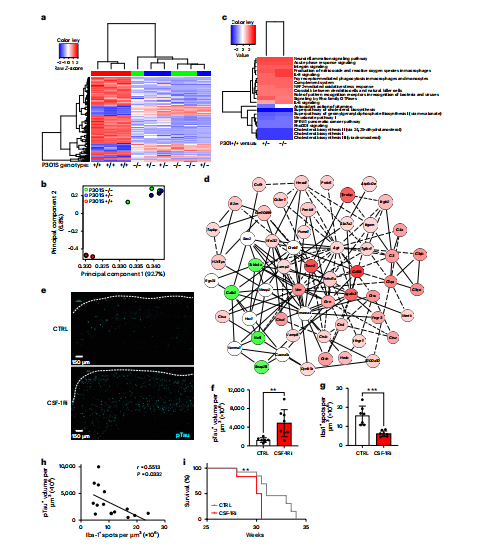
图2 CSF-1R抑制导致tau沉积增加和神经功能衰退
小胶质细胞作为大脑固有的免疫哨兵,是应对tau病理的第一道防线。为评估其功能贡献,研究者在疾病早期临床症状出现时,使用集落刺激因子1受体抑制剂PLX3397短期清除小胶质细胞。结果如图2e-i所示,清除小胶质细胞非但未缓解病情,反而导致大脑皮层内tau蛋白病理沉积显著增加,且小鼠的生存期明显缩短。该实验证实,在此疾病阶段,小胶质细胞整体上发挥着吞噬并限制异常tau蛋白播散的关键保护作用。然而,慢性、持续的病理刺激使小胶质细胞不堪重负。随着疾病进展,它们逐渐转变为一种高表达MHC II分子的活化状态,呈现出类似“抗原提呈细胞”的表型。这种长期处于激活状态的“痛苦”小胶质细胞,其功能可能从有益转向有害,构成了神经免疫稳态中的一个脆弱环节。
当机体先天免疫防线出现疲态时,适应性免疫系统启动了特异性应答。通过高分辨率多重免疫荧光技术,研究首次在图3中直观展示了这一细胞互作。在tau病变小鼠的脊髓(图3a, b)以及患有慢性创伤性脑病的人类患者脑组织tau病变区域(图3c),CD8+T细胞特异性地浸润并聚集,与Iba1+小胶质细胞形成了直接的、紧密的物理接触。量化分析表明,病变小鼠脊髓中约76%的CD8+T细胞处于这种接触状态。这证明,“CD8+T细胞-小胶质细胞”的相互作用是tau蛋白病中一种跨物种保守的普遍免疫特征。
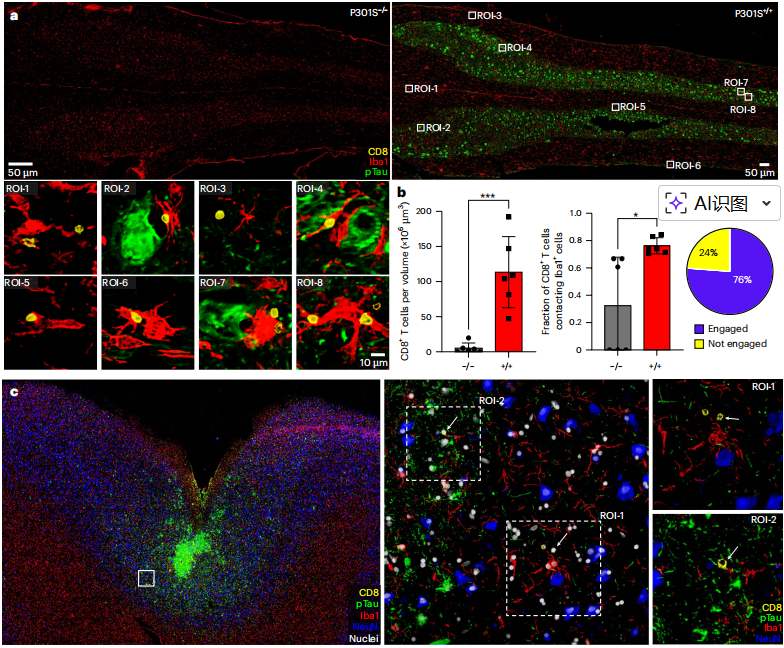
图3 CD8+T细胞在小鼠和人类tau蛋白病变中与小胶质细胞相互作用
为深入解析这群CD8+T细胞的性质,研究者进行了整合单细胞T细胞受体测序与转录组学分析。研究采用了AIMS生物信息学流程,基于T细胞受体互补决定区的生物物理特性对TCR序列进行聚类,成功在病变小鼠脊髓中鉴定出由克隆性扩增T细胞主导的TCR序列簇(如簇17、23)(图4a-c)。这些克隆细胞展现出高度一致的转录特征:它们不表达传统效应分子如干扰素-γ、肿瘤坏死因子或颗粒酶A/B/C,却特异性高表达颗粒酶K,并共表达免疫检查点分子TIGIT和PD-1(图4d-g)。这定义了一类新型的、处于受调节状态的效应T细胞亚群。
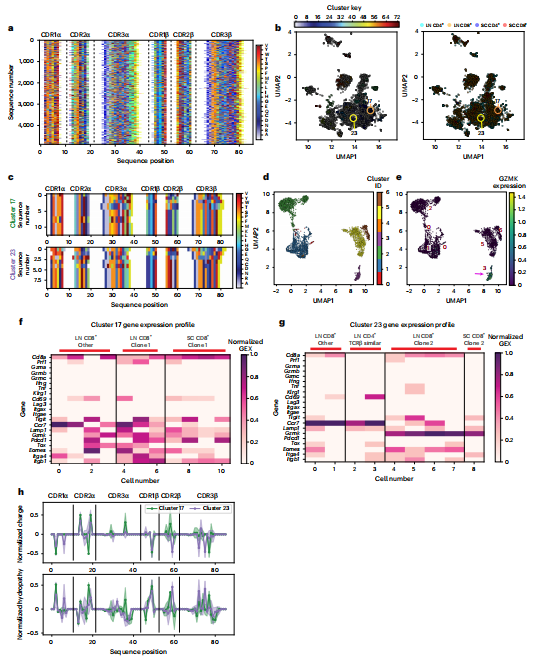
图4 自动免疫分子分离(AIMS)分析鉴定出具有相似转录特征、生物物理特性相近的TCR簇
随后,研究者采用单细胞RNA测序将脊髓中的CD8+T细胞划分为数个亚群,其中簇3几乎独占性地高表达GzmK基因(图5c-e)。对该簇的深入分析(图5f, g)揭示了其完整的分子特征:这是一群同时表达归巢受体、效应相关分子及免疫调节蛋白的组织驻留样细胞毒性T细胞。系统发生树分析(图5a, b)进一步显示,存在多个克隆扩增的CD8+T细胞系,且在脊髓和引流淋巴结中均有分布,证实了这是一场抗原驱动的全身性特异性免疫应答。
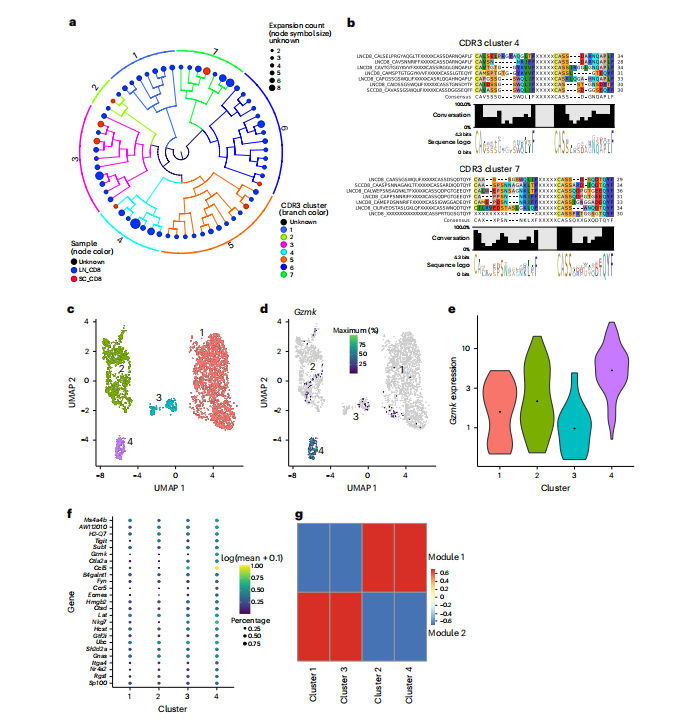
图5 单细胞RNA测序揭示克隆扩增的CD8⁺T细胞表达颗粒酶K(GZMK)
在小鼠脊髓的高倍成像中(图6a-e),可以清晰地看到GZMK+CD8+T细胞不仅接触小胶质细胞,更将GZMK蛋白直接沉积在靶细胞的表面。量化显示,病变环境中约78%的CD8+T细胞为GZMK阳性。在人类大脑的tau病变区(来源于慢性创伤性脑病、阿尔茨海默病及年龄相关病变患者)(图6f-h),研究者观察到了完全一致的景象:GZMK+CD8+ T细胞特异性定位于病变区域,与小胶质细胞形成免疫突触,并沉积GZMK。平均81%的病变区内CD8+T细胞表达GZMK。
至此,完整的证据链形成:在tau蛋白病中,存在一支抗原特异性、克隆性扩增的GZMK+CD8+T细胞亚群。它们被精确招募至病变部位,通过与功能失调的小胶质细胞形成免疫突触并沉积GZMK效应分子来行使功能。
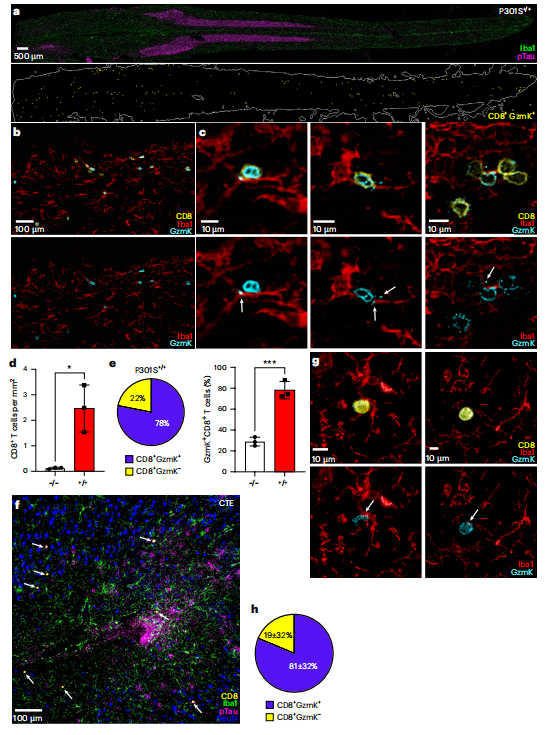
图6 GZMK⁺CD8⁺T细胞在小鼠和人类tau病变中靶向小胶质细胞
为确证这支“特警”细胞的保护性功能,研究者构建了CD8+T细胞缺陷的tau病变小鼠模型。细胞缺失导致了全面的病理加重:大脑内tau蛋白负荷显著增加,外周血中反映轴突损伤与神经胶质激活的生物标志物水平升高(图7a-c)。脊髓中高度活化的CD11c+MHCII+小胶质细胞异常增多,并出现了一群多核的、含有神经元来源转录本的异常小胶质细胞亚群(图7d, e; 图8a-e)。单细胞测序显示此亚群仅存在于CD8+T细胞缺陷鼠中,并高表达抗病毒相关基因。此外,行为学测试表明,缺陷小鼠的运动功能衰退速率显著加快(图8f)。这些反向遗传学实验证明,GZMK+CD8+T细胞的核心生理功能是清除或抑制那些功能失调、可能有害的小胶质细胞,从而维持大脑免疫微环境的稳态,最终延缓疾病整体进程。
任何有效的免疫反应都需要内在的制衡机制。研究发现,这群保护性T细胞自身高表达免疫检查点蛋白PD-1和TIGIT。当使用中和性抗体阻断这两种抑制性信号时,尽管CD8+T细胞总数未变,但其在脊髓灰质中的分布发生改变,多核小胶质细胞簇增多,并最终加速了小鼠的神经功能衰退(图8g-k)。这表明,PD-1/TIGIT信号通路是维持这种保护性免疫反应适度、可控,防止其过度激活造成“旁观者损伤”的关键安全阀。
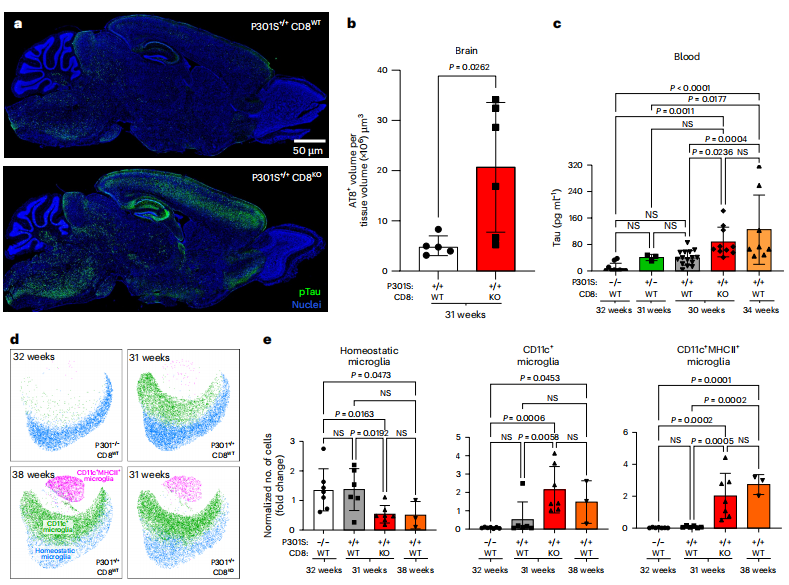
图7 CD8⁺T细胞降低pTau病理与高度活化的小胶质细胞
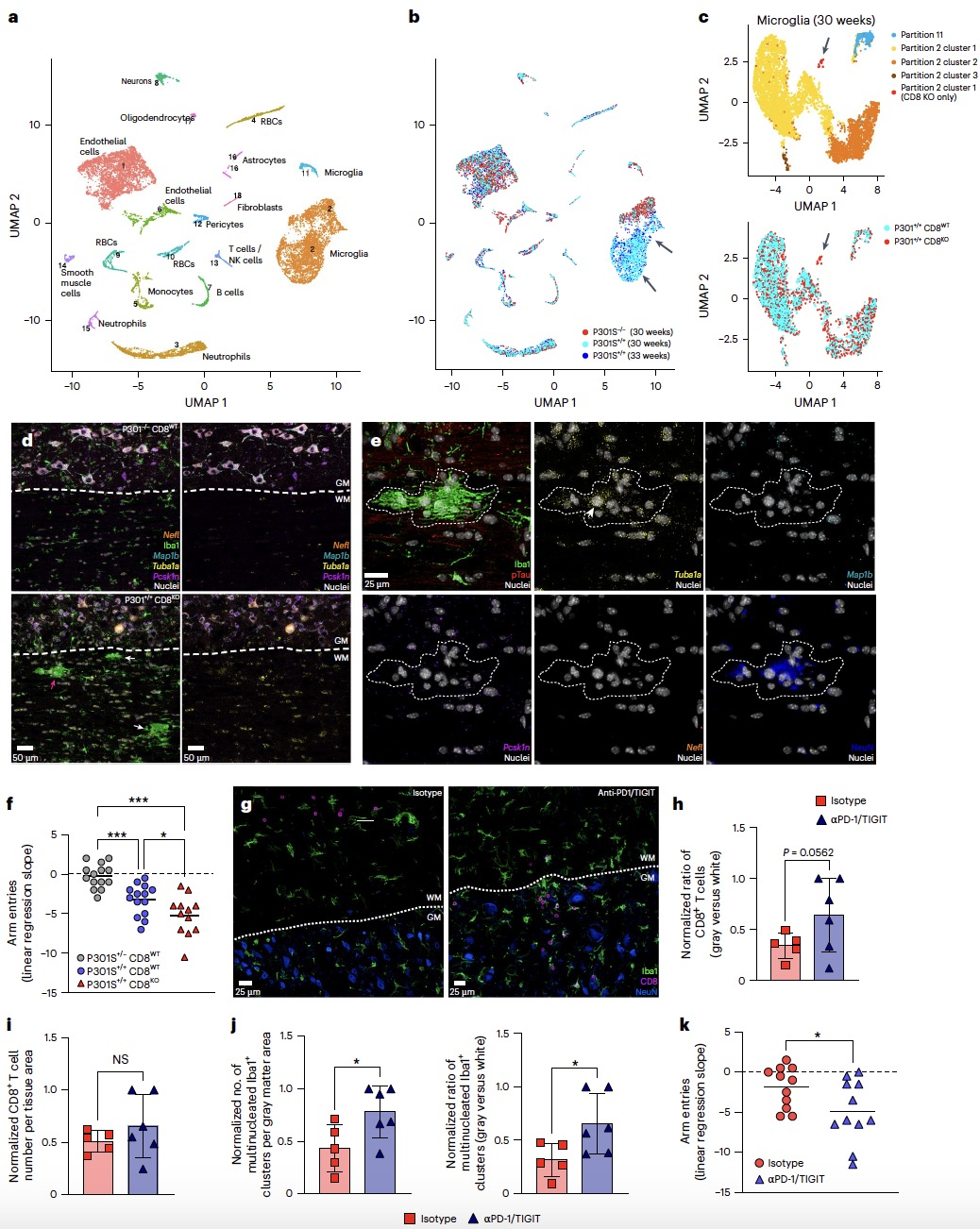
图8 CD8⁺T细胞清除携带神经转录本的小胶质细胞并受PD-1/TIGIT调控
醉翁之艺点评
本研究构建了一个全新的、动态的“免疫协同防御”模型来阐释tau蛋白病的进展:小胶质细胞作为先天免疫主力,在疾病早期阶段通过吞噬作用限制tau蛋白的初始播散。随着疾病的进展, GZMK+ CD8+ T细胞作为适应性免疫的精确打击力量,被抗原特异性激活并克隆扩增,进而识别、清除或重置功能失调的小胶质细胞,防止其转为致病因素。其中,PD-1/TIGIT等免疫检查点作为反馈调节枢纽,确保整个防御反应精准、适度,避免免疫病理损伤。
本研究将免疫系统在神经退行性疾病中的作用,从既往相对模糊的“炎症破坏者”角色,精确定义为一个具有“监视、维护与更新”功能的精密调节网络,深化了对神经-免疫环路互作的理解。GZMK+ CD8+ T细胞及其调控通路(如TIGIT/PD-1)成为极具潜力的治疗干预靶标。未来的治疗策略可能着眼于“精准调变”而非“广泛抑制”免疫反应,例如开发旨在温和增强该特定T细胞亚群功能或解除其不当抑制的药物。同时,该研究也提示了在神经退行性疾病患者中应用免疫检查点抑制剂时,必须高度谨慎并进行充分评估,因为盲目增强T细胞效应功能可能破坏这种保护性平衡,导致病情意外加速。