深度解析医学证据,DeepEvidence为你支撑决策
前言:2026年3月,上海交通大学第六人民医院贾伟教授、郑晓皎研究员团队在国际代谢顶刊《Cell Metabolism》在线发表重磅研究,首次系统阐明过量糖摄入→肠道菌群乙醛发酵→内源性乙醛→MMP7驱动肝纤维化的完整因果链,并构建高效乙醛清除工程化益生菌,为MASLD-MASH微生态靶向治疗提供全新策略。
该研究基于UK Biobank超21万人群队列分析,结合多组学技术和临床前模型,首次系统阐明了过量糖摄入通过肠道菌群发酵产生内源性乙醛驱动MASLD向MASH进展的完整机制链,并成功构建具有高效乙醛清除能力的工程化益生菌Ligilactobacillus salivarius HAM,在动物模型中显著阻断肝纤维化进程。该研究为MASH的微生物组靶向治疗提供了全新的精准干预靶点和概念验证。
1 研究背景
领域核心痛点:代谢相关脂肪性肝炎(MASH)是MASLD进展的关键节点,可致肝硬化、肝癌,尚无有效阻断手段。过量果糖与MASLD密切相关,但肠道菌群源性内源性乙醛在非酒精性肝纤维化中的作用与机制,长期未被系统解析。
在生活方式因素中,糖摄入(尤其是果糖)与MASLD发病密切相关,但其加速MASLD向MASH进展的具体机制尚不清楚。近年来,肠道微生态失衡及其代谢产物在MASLD发病中的作用日益受到重视。值得关注的是,与酒精性肝病(ALD)中外源性酒精摄入不同,肠道菌群产生的内源性酒精和醛类物质在MASLD-MASH进展中的贡献尚未被系统阐明。越来越多的观察性研究发现MASH患者循环中内源性醇类水平升高,这提示微生物代谢可能参与疾病进展,但具体的分子机制和因果关系仍有待揭示。
2 研究设计与方法
本研究采用了“临床大队列分析—多组学机制探索—功能验证—治疗策略开发”的整合研究框架:
(1)流行病学分析:利用UK Biobank数据库210,861名参与者的饮食摄入与临床结局数据,通过广义可加模型(GAM)和Cox比例风险模型分析糖摄入与肝病进展及死亡的关联。
(2)多组学机制研究:对欧洲NAFLD注册队列146例经活检确诊的MASLD/MASH患者进行粪便宏基因组和宏转录组测序;同时对25例临床队列(健康对照、MASLD、MASH各5、8、12例)进行粪便体外转化实验和代谢物定量分析。
(3)动物与细胞模型验证:构建高脂饮食(HFD)及高脂高果糖饮食(HFHFD)小鼠模型,通过抗生素干预、乙醛补充、基因敲除(全身Aldh2 KO、肝特异性Aldh2 KD、HSC特异性Mmp7 KD)等多种策略验证因果关系。采用RNA-seq、细胞类型定位、Western blot等技术解析分子机制。
(4)工程化益生菌开发与评价:从51例长期饮酒但无显著肝纤维化(FIB-4<1.45)的ALD患者粪便中筛选高效乙醛代谢菌株,通过全基因组测序鉴定并构建过表达双功能乙醛-醇脱氢酶(adhE)的工程菌L. salivarius HAM,在HFHFD小鼠模型中评估其治疗效应及28天亚急性毒性。
主要评价终点包括:肝组织病理学(H&E、Sirius Red染色)、NAFLD活动度评分(NAS)、血清ALT/AST、肝/粪便乙醛水平、纤维化相关基因表达等。
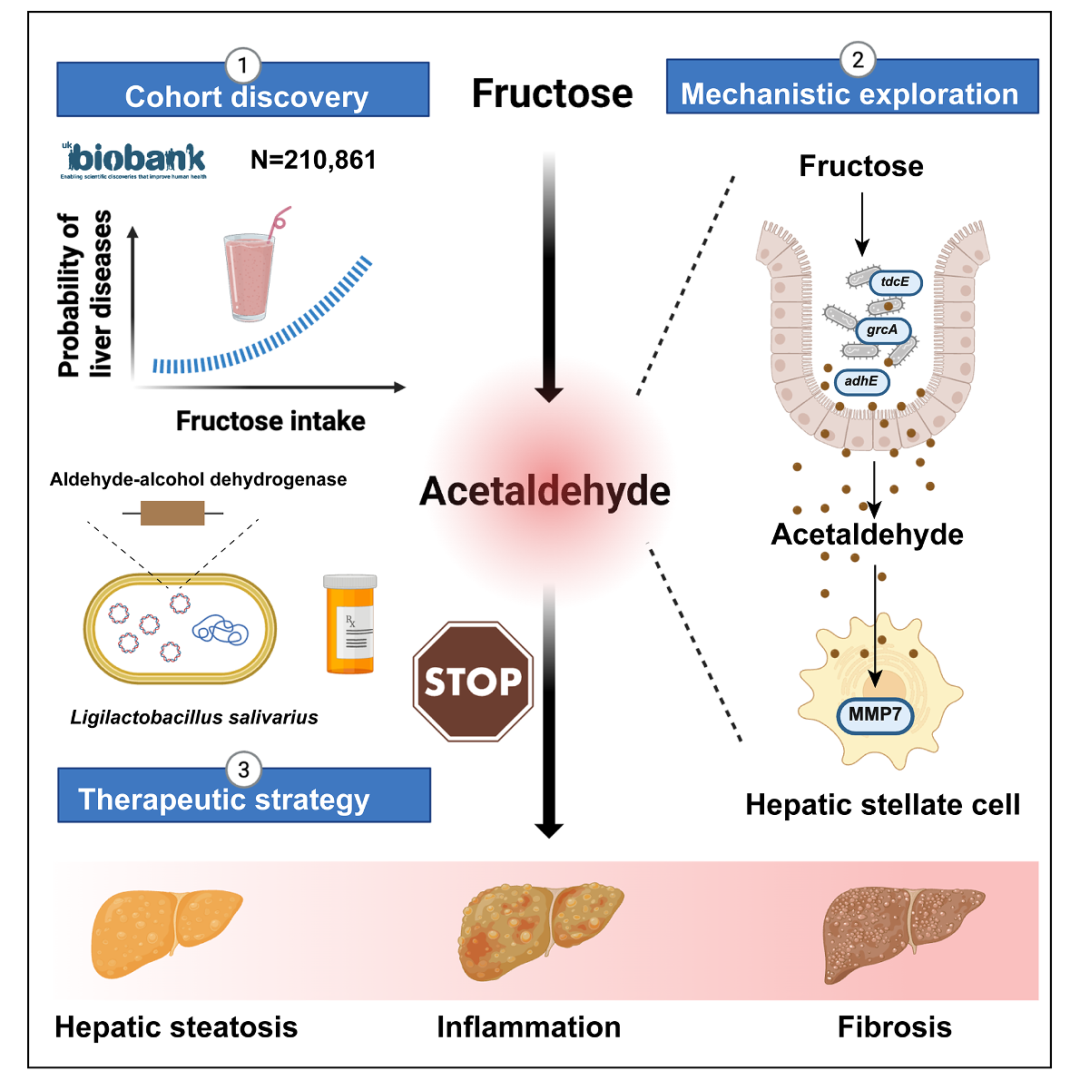
图1:研究机制概览——从果糖摄入到肠道菌群乙醛发酵,经MMP7驱动肝纤维化,再到工程化益生菌精准干预的完整通路
3 研究结果
3.1 糖摄入与MASLD-MASH进展:超大队列的剂量-反应关系
UK Biobank分析揭示了令人警醒的剂量依赖性关联:总糖摄入与全因死亡率呈非线性J型关系,且与肝病发病率呈显著正相关。将糖摄入按四分位分组后,最高摄入组(Q4, >154.9g/d)的10年肝脏相关生存率显著低于最低摄入组(Q1, <88.0g/d)。Cox回归分析进一步显示,在各类糖中,果糖的肝脏相关死亡风险升高最为显著(HR=1.86,95% CI 1.66–2.08)。GAM分析调整了年龄、性别、BMI和吸烟状态后,上述关联依然稳健。
在动物模型中,HFHFD喂养小鼠的纤维化进展远超单纯HFD组:18周即出现早期纤维化,28周形成进展期桥接纤维化(纤维化评分2分,胶原面积5.92%→8.63%);而HFD组至56周纤维化仍较轻微。标准化的60%高脂+10%果糖水验证实验同样证实,果糖补充显著加重脂肪变并加速纤维化。
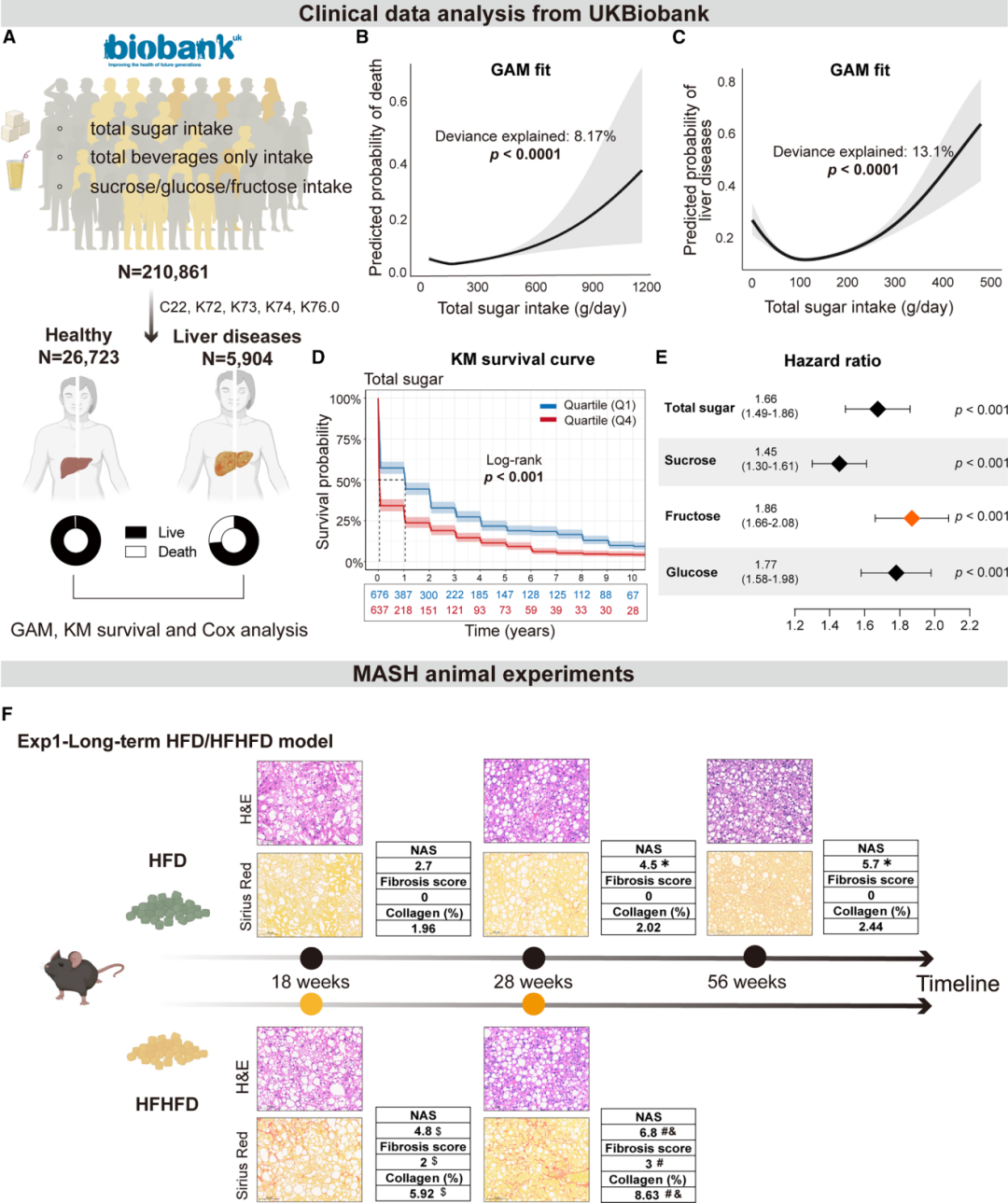
图2:UK Biobank队列中糖摄入与肝病死亡/发病的剂量-反应关系(上),以及HFHFD加速小鼠肝纤维化进展的组织学证据(下)
3.2 肠道菌群乙醛发酵通路激活:从相关性到因果性的关键证据
宏基因组通路分析揭示,MASH患者肠道微生物的糖酵解下游通路——丙酮酸发酵生成乙醛/乙醇通路(MetaCyc: PWY-5480)在MASH阶段显著富集,随机森林模型将其识别为区分MASH与早期阶段的最具判别力的通路之一。PCoA分析显示MASLD、MASH-0和MASH-1三组的菌群结构明显分离。
关键的功能验证来自抗生素干预实验:广谱抗生素清除肠道菌群后,HFHFD小鼠的桥接纤维化几乎完全消失,NAS从4.8降至3.0,纤维化评分从2分降至0分。这直接证明菌群是糖诱导MASLD-MASH进展的必需媒介。
粪便体外转化实验提供了更进一步的功能证据:MASH患者粪便在果糖孵育下产生的乙醛量是MASLD患者的3.8倍(葡萄糖条件下为3.9倍);HFHFD小鼠粪便产乙醛量分别为HFD小鼠的2.4倍(果糖)和1.6倍(葡萄糖)。纵向监测显示,HFHFD小鼠粪便乙醛在第3周即显著升高并持续积累,且与NAS评分呈显著正相关(R=0.86,P<0.001)。在临床队列中,MASH患者粪便乙醛水平显著高于MASLD患者,并与FIB-4指数呈正相关(R=0.84,P<0.01)。
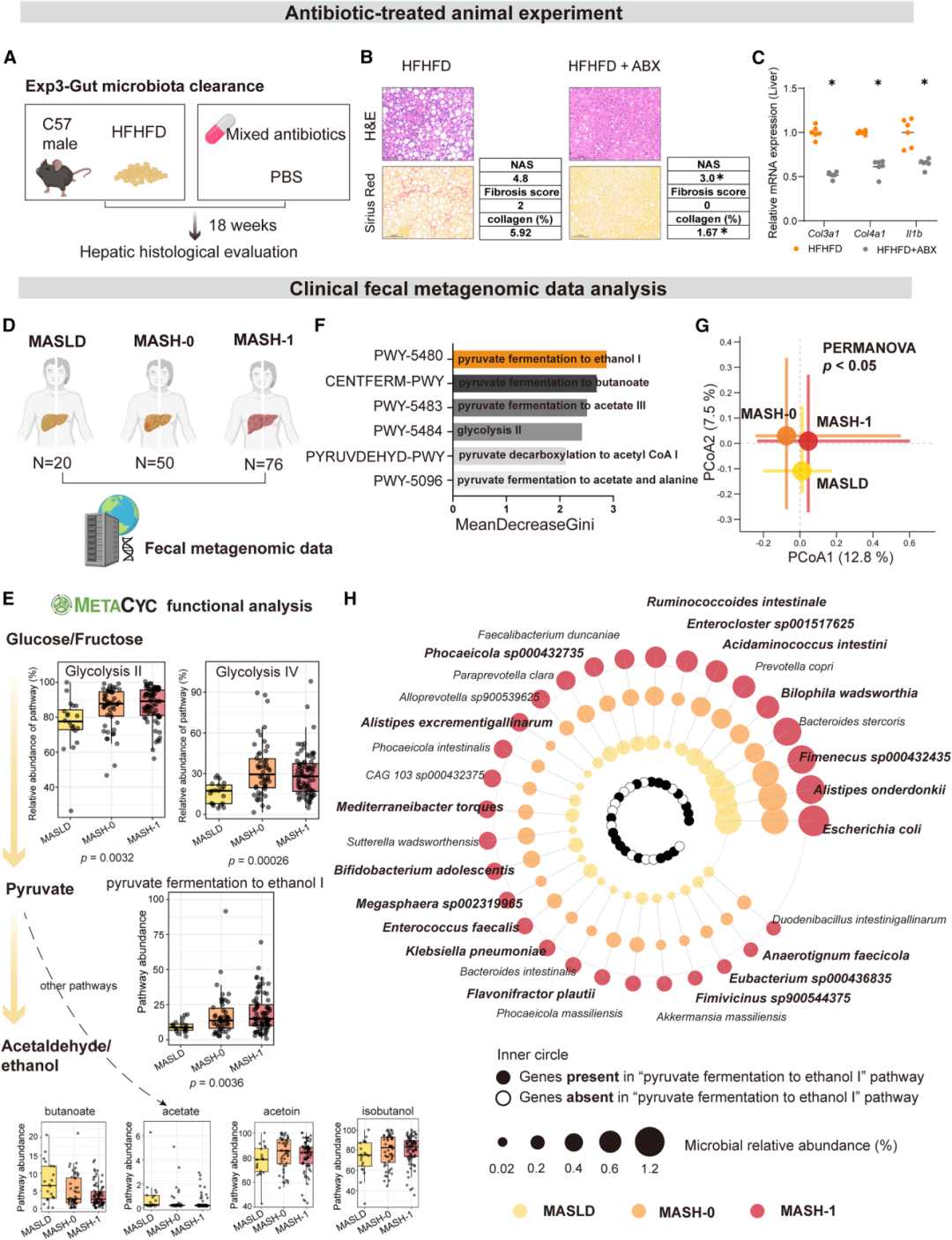
图3:HFHFD+抗生素干预证实菌群必需性(上);临床宏基因组分析揭示MASH中乙醛发酵通路(PWY-5480)显著富集(下)
3.3 内源性乙醛的微生物起源:排除宿主代谢通路的干扰
研究通过精巧的实验设计,确证了乙醛的微生物来源。尽管乙醛沿肠-肝轴同步升高(小肠内容物、远端小肠、结肠、门静脉血、肝脏均显著升高),但关键发现是:各compartment中乙醇浓度并未伴随乙醛升高而增加,两者出现“解耦”。宏转录组分析显示,MASH患者中驱动丙酮酸→乙醛通路的关键基因(tdcE、grcA、adhE)转录丰度显著升高。
为排除宿主代谢的贡献,研究团队进行了两项关键干预:使用宿主ADH抑制剂4-甲基吡唑(4-MP)虽显著升高乙醇水平,却未能降低肝脏乙醛含量;而全身Aldh2基因敲除小鼠肝脏乙醛进一步飙升(约1.7倍),乙醇仍无变化。肠道和肝脏组织中Adh1和Aldh2的表达在两组间亦无差异。上述证据链明确排除了宿主乙醇氧化通路的贡献,指向过量乙醛主要来源于肠道菌群的丙酮酸发酵通路。
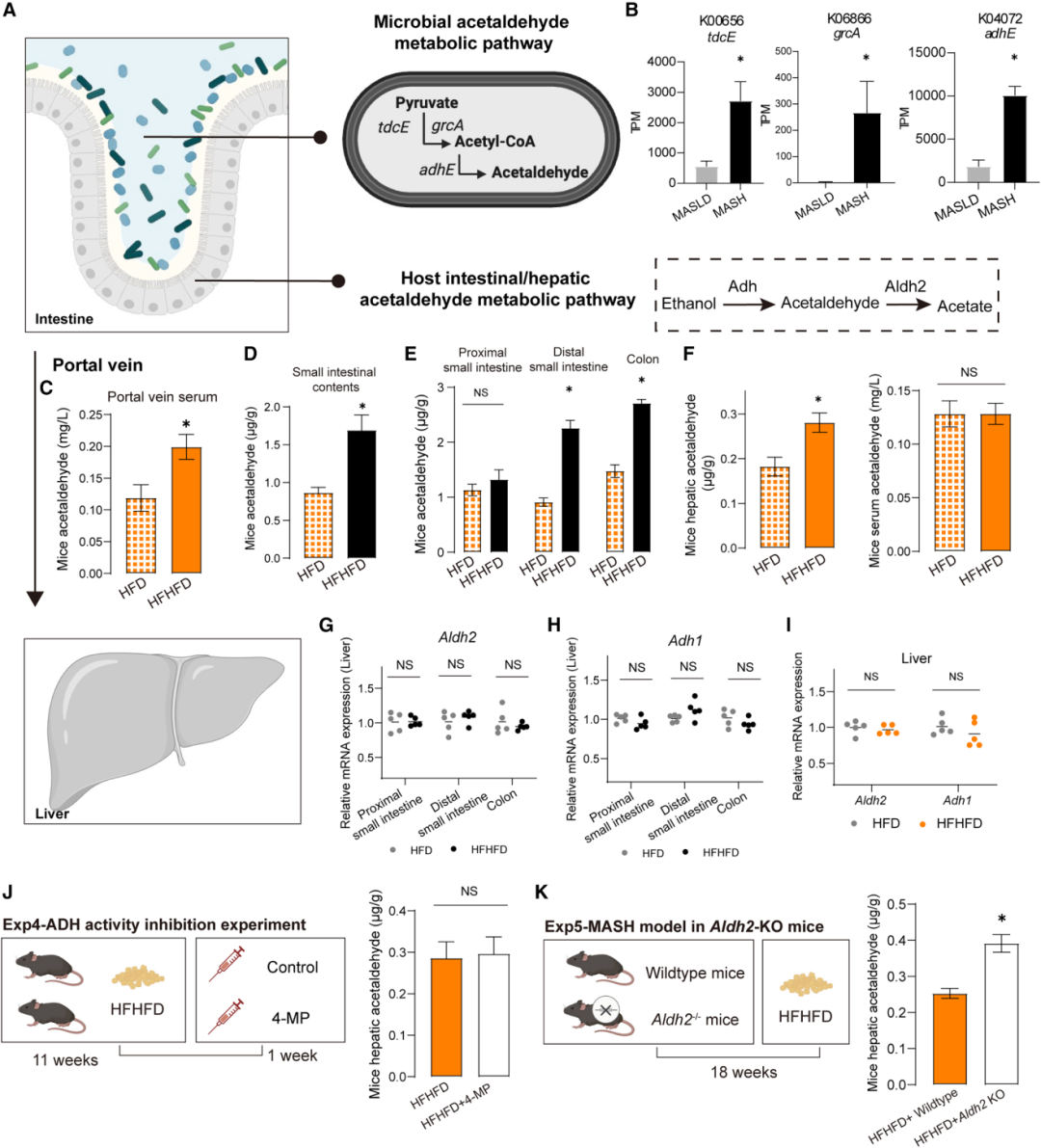
图4:微生物与宿主乙醛代谢通路对比(上);宏转录组证实MASH中关键基因(tdcE、grcA、adhE)显著上调(右上);肠-肝轴乙醛定量分布及ADH/ALDH2实验排除宿主来源(下)
3.4 乙醛-MMP7-肝星状细胞轴:纤维化驱动的新机制
低剂量乙醛(50mg/kg/d)口服干预18周HFD小鼠,显著加重肝脏炎症和胶原沉积(纤维化评分从0升至2分,胶原从1.96%升至4.25%),促炎基因(Tnfa、Il1b)和促纤维化基因(Col1a1、Col3a1、Tgfb、Timp1)表达上调。
RNA-seq交叉比较识别出730个共有差异表达基因,GO富集指向胶原代谢和细胞外基质(ECM)组织通路,细胞类型定位分析将肝星状细胞(HSCs)锁定为关键效应细胞——49个差异基因富集于HSCs,远超Kupffer细胞(20个)、内皮细胞(18个)和肝细胞(10个)。
机制上,乙醛在LX2人肝星状细胞中显著上调基质金属蛋白酶-7(MMP7)的表达。Western blot证实乙醛干预小鼠肝脏和LX2细胞中MMP7蛋白水平均升高;公共数据集(GDS4881)显示MASH患者MMP7 mRNA较健康对照升高约8倍。功能实验证实,MMP7抑制剂可显著削弱乙醛诱导的纤维化基因表达;更关键的是,利用HSC特异性GfaABC1D启动子驱动的AAV-shMmp7敲低,在HFHFD条件下显著改善肝纤维化(纤维化评分从0升至1分,胶原从1.05%升至3.50%),从因果层面确立了MMP7在这一通路中的核心介导作用。
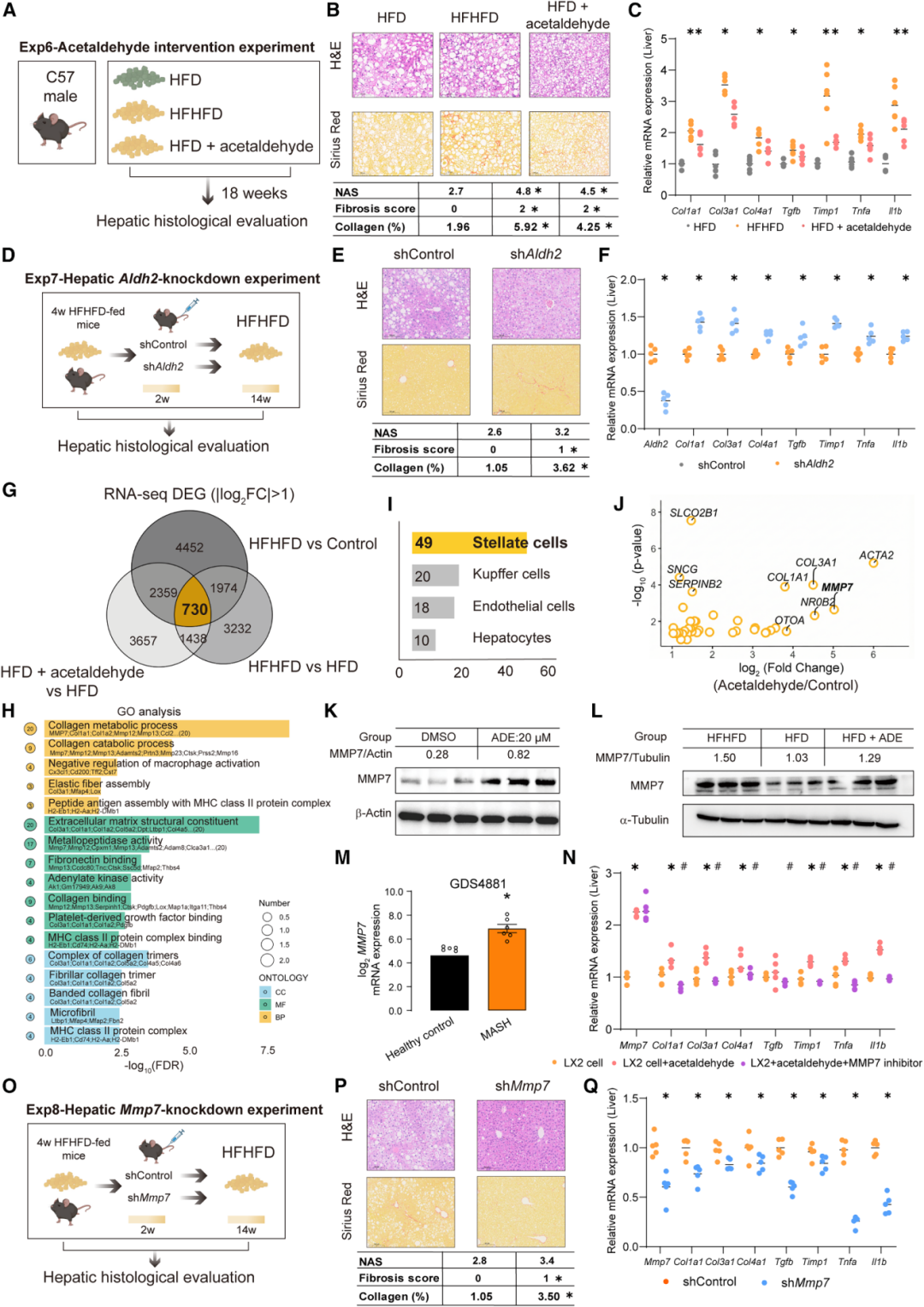
图5:乙醛干预及肝Aldh2敲低加重纤维化(A-F);RNA-seq交叉分析识别730个DEG并定位HSC为关键效应细胞(G-J);MMP7在乙醛诱导纤维化中的因果作用验证(K-Q)
3.5 工程化益生菌L. salivarius HAM:精准清除乙醛的微生物组疗法
从健康饮酒者粪便中筛选获得的L. salivarius野生株即表现出卓越的乙醛清除活性(较标准菌株ATCC 11741提高10.9倍),经adhE基因工程改造后的L. salivarius HAM株更是将乙醛清除能力提升了34.9倍(乙醇+乙醛条件下)。
治疗性给药实验令人振奋:HFHFD小鼠经L. salivarius HAM干预6周后,血清ALT从~280U/L降至~230U/L、AST同步下降,肝脏炎症、脂肪变和纤维化明显改善——NAS从6.8降至4.2,纤维化评分从3分降至0分,胶原面积从8.63%降至1.92%;肝脏和粪便乙醛水平均大幅下降,肝内Mmp7及促炎/促纤维化基因表达显著下调,且效果优于野生株和标准株。28周长期给药同样证实获益持续。Western blot显示HAM组MMP7蛋白表达较PBS组显著降低(MMP7/Tubulin比值从1.49降至0.93)。宏基因组分析显示益生菌治疗对肠道菌群整体多样性影响甚微,提示其通过靶向代谢而非重塑菌群发挥治疗作用。
安全性方面,28天亚急性毒性评估(高剂量2×10¹⁰CFU/只)显示心、肝、脾、肺、肾、肠等主要器官H&E染色无异常,血清ALT、AST、TBIL、CREA均在正常范围,证实良好的生物安全性。
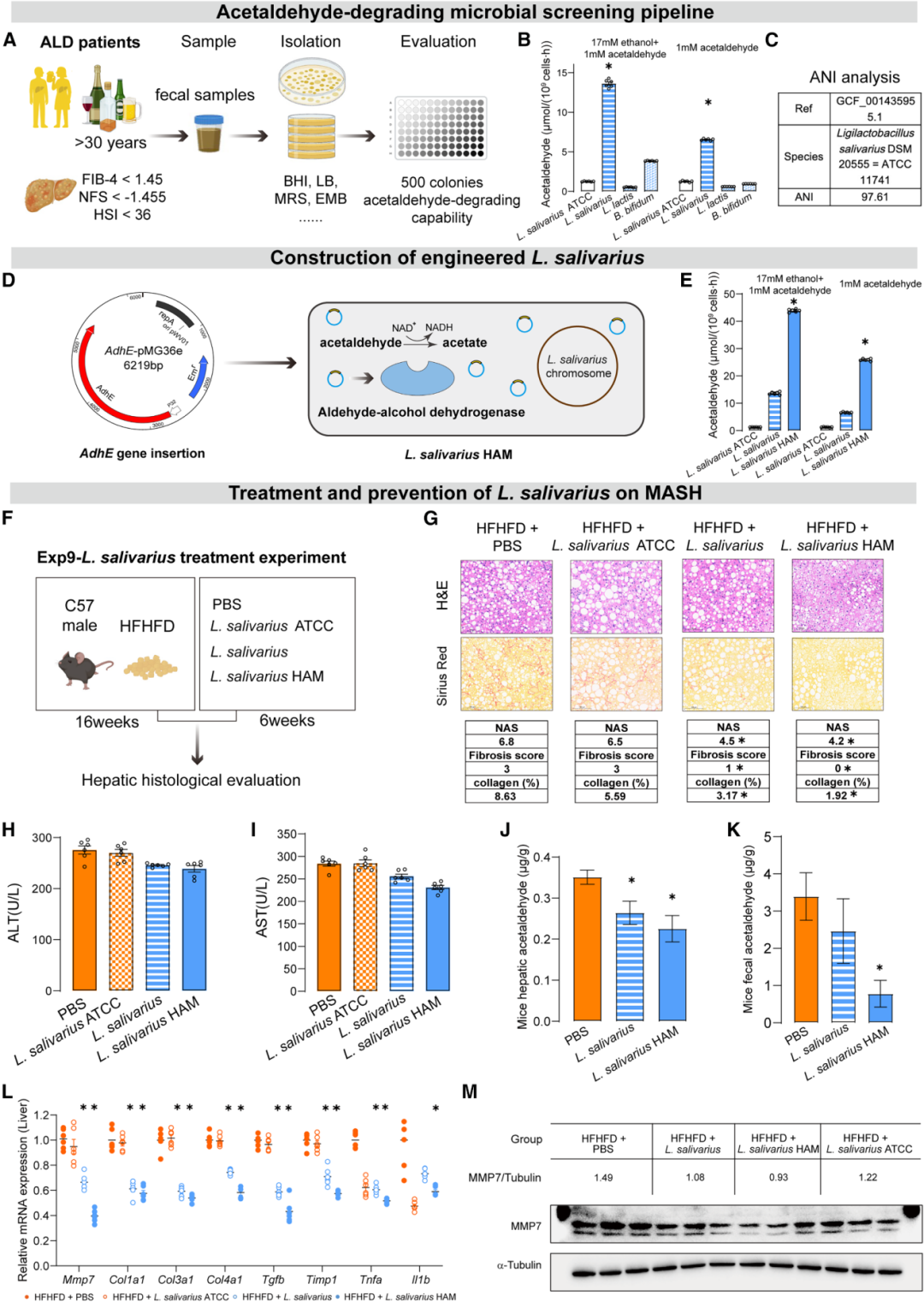
图6:从ALD患者粪便筛选高效乙醛代谢益生菌(上);L. salivarius HAM工程化构建使乙醛清除能力提升34.9倍(中);益生菌治疗显著改善HFHFD小鼠肝脏组织学、降低乙醛水平并下调MMP7表达(下)
4 研究意义
这项研究在MASLD-MASH的基础与转化研究领域具有多重里程碑意义:
(1)揭示糖-菌-肝轴的全新致病机制:首次建立“高糖摄入→肠道菌群乙醛发酵通路激活→内源性乙醛产生→HSC中MMP7上调→肝纤维化”的完整因果链,填补了内源性微生物代谢物在MASH进展中作用的研究空白,为理解饮食-微生态-肝脏交互作用提供了新范式。
(2)确立内源性乙醛作为潜在无创标志物:粪便乙醛水平与NAS、FIB-4等纤维化指标显著正相关,且可在疾病早期(小鼠第3周)即出现升高,具备作为MASH进展风险分层标志物的潜力。
(3)开创精准微生态治疗新策略:不同于传统的抗炎或抗氧化益生菌作用模式,本研究首次采用“代谢功能增强”的工程化益生菌策略,通过定向强化特定致病代谢物的清除能力来阻断纤维化,代表了微生物组疗法从“菌群重塑”向“代谢精准干预”的范式转变,为MASH的防治提供了极具临床转化前景的新型治疗手段。
(4)连接更广泛的醛类代谢疾病谱:研究将MASH中的微生物乙醛代谢与ALDH2*2突变相关的多种疾病(阿尔茨海默病、糖尿病、心血管疾病等)在机制上建立了联系,提示肠道菌群源性醛类代谢可能是多种代谢性疾病的共同病理基础。
5 总结与展望
本研究从流行病学关联出发,经由多组学机制解析、多层次因果关系验证,最终走向治疗策略的概念验证,形成了MASLD-MASH研究中“机制-靶点-干预”的完整闭环。核心结论清晰有力:过量糖摄入(特别是果糖)通过重塑肠道菌群代谢功能,驱动内源性乙醛的微生物发酵产生,后者经肠-肝轴转运至肝脏,激活HSC并上调MMP7表达,从而促进纤维化;靶向清除肠腔乙醛的工程化益生菌可有效阻断这一进程。
当然,研究也存在一定局限性:代谢组学分析主要聚焦于乙醛这一代表性醛类,微生物糖发酵产生的其他潜在致病醛类物质尚未全面筛选;工程化益生菌的长期稳定性、肠道定植动态及在复杂人体环境中的安全性和有效性仍需系统评估。未来,期待在更大规模的纵向临床队列中验证粪便乙醛作为MASH无创标志物的诊断效能,并推动工程化益生菌向临床试验转化。随着对糖-菌-肝轴认识的不断深入,我们有望迎来MASH治疗从“被动管理”到“主动预防”、从“全身用药”到“肠道精准干预”的新阶段。
参考文献
Tang Y, Kuang J, Xia X, et al. Targeting microbiota-generated acetaldehyde to prevent progression of metabolic dysfunction-associated steatotic liver disease. Cell Metab. 2026;38(6):1-15. doi:10.1016/j.cmet.2026.01.021.