首页 > 医疗监管/ 正文
深度解析医学证据,lxfs.net为你支撑决策
在全球范围内,罕见遗传疾病的发生率约为每 1000 例活产婴儿中有 40-82 例。明确罕见遗传病的分子致病病因,有助于实现早期确诊、优化诊疗方案制定以及开展精准遗传咨询。马来西亚总人口 3400 万,预估 150 万~200 万(占比 4%~6%)人口罹患罕见病,其中绝大多数为遗传性罕见病。马来西亚遗传门诊接诊患者可开展的检测项目包括常规核型分析、脆性 X 综合征检测、代谢筛查以及单基因专项检测。受实验室资源短缺限制,染色体微阵列分析(CMA)与基因panel检测无法作为常规检查开展,因此绝大多数罕见病患者常年无法确诊。全外显子测序(WES)是一项综合性检测手段,既可检出染色体微阵列分析(CMA)所能发现的大片段基因缺失与重复变异,也能识别各类基因panel筛查可检出的基因变异。现阶段 WES 具备良好成本效益,在全球范围内正逐步成为疑似罕见遗传病患者的临床常规检测项目。本研究为单中心回顾性研究,研究地点设于马来西亚当地一家遗传诊疗中心。2020 年 8 月至 2021 年 12 月期间,经临床遗传专科医师临床评估后,对所有疑似遗传性疾病的先证者开展仅检测先证者样本的 WES 检测;测序实验在韩国一家权威参比实验室完成。从病例档案中收集患者人口学资料、临床表型、基因检测结果并开展统计分析。
总计 489 名患者完成 WES 检测,其中 83%(407/489)为儿科病患;大部分患者以神经发育异常为主要就诊表现。WES在 50%(243/489,95%置信区间(CI):45%-54%)的患者中确定了潜在的基因病因。在补充新增临床表型信息或无额外临床资料的前提下,对初次阴性的 228 例样本重新分析 WES 数据,额外确诊 17 例患者,最终整体确诊率达 53%(260/489,95%CI:49%~58%)。本研究中有 9 例患者检出双重分子基因学致病诊断结果,21 例患者确诊存在拷贝数变异(CNV)。已确诊病例的遗传方式分布:常染色体显性遗传占 64%(171/268)、常染色体隐性遗传占 24%(65/268)、X 连锁遗传占 12%(32/268)。82%(401/489)的患者选择接受次要发现(在初始结果为阴性或不确定的情况下,允许他们决定是否接受次要发现,并定期重新分析他们的WES数据。),其中 5.5%(22/401,95%CI:3.6%~8.2%)的受检者检出致病性/疑似致病性(P/LP)变异,相关变异基因分别关联心血管疾病(占 64%,14/22)或肿瘤疾病(占 27%,6/22)。
本研究证实,WES 能够高效帮助马来西亚遗传门诊就诊的疑似罕见遗传病患者明确分子诊断。遵照 2019 年联合国相关政治宣言内容,筹措专项经费以保障 WES 检测落地,是马来西亚实现罕见病患者全民普惠医疗目标的关键举措。
研究背景
全球范围内,罕见遗传病在新生儿中的发生率为每千活产儿 40~82 例。明确罕见遗传病的分子发病机制,能够帮助患者尽早确诊,缩短罕见病患者普遍经历的漫长 “确诊漂泊期”。所谓确诊漂泊期,即患者从初次出现临床症状直至获得精准疾病诊断的整个漫长就医历程,漫长的确诊周期会给患者及其家属带来巨大心理压力与精神困扰。获得确切的分子诊断结果,同样能够指导临床制定诊疗方案,包括并发症定期监测与靶向药物治疗。精准的基因诊断是评估子代再发风险、开展遗传咨询的先决条件,帮助患者与家属从容面对遗传性疾病。除此之外,依托精准分子诊断,研究者可汇总多中心病例数据,系统完善各类罕见病自然病程与预后相关临床资料。
多年以来,基因检测技术与手段持续迭代升级。依托二代测序(NGS)技术发展,全外显子 / 全基因组测序(WES/WGS)可对全部蛋白编码区或人类全基因组进行测序,正逐步纳入临床常规检测项目。这类基因组测序技术除检出各类小基因变异外,还可替代核型分析、染色体微阵列分析(CMA)检出染色体结构变异,因此其确诊阳性率显著高于核型分析(5%~10%)以及染色体微阵列分析(CMA,15%~20%)。针对疑似遗传性疾病的患者群体,WES 临床确诊率区间可达 25%~58%。同时,WES 无需临床医师依据患者临床表现预先锁定可疑致病基因再分批测序,避免患者依次接受多项单基因或基因包序贯检测,大幅缩短确诊耗时,对于临床表型高度异质的遗传性疾病尤为适用。但与此同时,意义未明变异(VUS)以及次要分析结果往往需要患者家属补充家系验证检测,可能为患者带来心理焦虑、经济与医疗负担。
在马来西亚,遗传门诊可开展的遗传病检测项目十分有限,仅包含传统染色体核型分析、选择性荧光原位杂交(FISH)检测、遗传性代谢病生化筛查、脆性 X 综合征检测以及数十项基于Sanger测序的单基因检测。受实验室资源匮乏限制,染色体微阵列分析(CMA)无法常规开展;商业化实验室提供的二代测序相关检测(多基因panel检测、WES/WGS)仅可供自费患者选用。以上现状造成大量患者长年无法确诊,临床疑诊难以通过基因检测得到验证。本文汇报一项在马来西亚马来半岛北部区域性遗传诊疗中心开展的 WES 临床研究,该中心服务覆盖约 650 万人口,也是马来西亚迄今为止样本量最大的 WES 队列研究。
研究结果
确诊率数据
2020 年 8 月至 2021 年 12 月共计 17 个月内,489 名受试者完成先证者 WES 测序。489 例中 83%(407/489)为 18 周岁以下儿科患者,剩余 17% 为成年受试者;407 例儿科患者里,新生儿占 3%(14 例),全部受试者均仅开展先证者单独测序。
33.3%(163/489)受试者检出致病性或可能致病性(P/LP)变异,结果阳性;20%(98/489)结果存疑,包含常染色体显性致病基因检出单个杂合意义未明变异(VUS)、常染色体隐性致病基因检出复合杂合 / 纯合 VUS、常染色体隐性致病基因检出单个 P/LP 变异三类情况;剩余 46.7%(228/489)初次测序阴性。98 例结果不确定受试者中,经致病机制研讨与变异家系分离验证后,80 例可与临床表现形成明确临床关联并纳入确诊病例,至此 WES 初始确诊率达 50%(243/489,95% CI:45%~54%)。依托新增临床表型资料或仅通过原有测序数据重新分析 WES 数据后,额外 17 例初筛阴性患者因新发致病基因研究进展、VUS 重新定级为 P/LP 获得确诊(图 1),研究整体最终确诊率 53%(260/489,95% CI:49%~58%)。
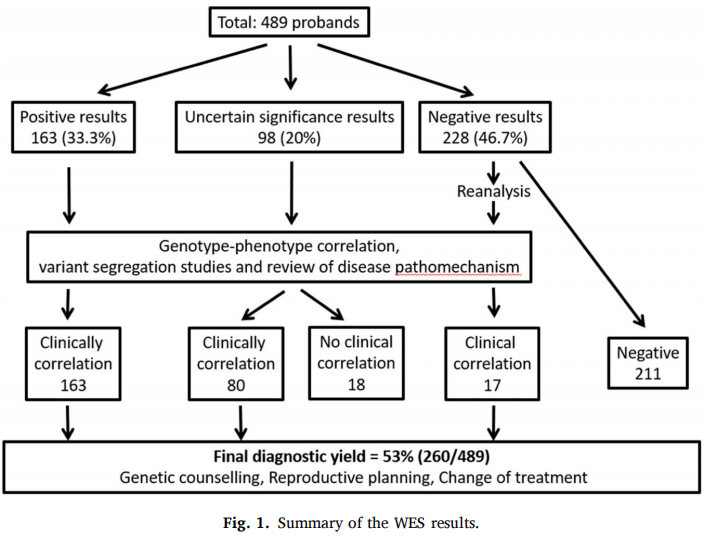
图1
260 例确诊病例中共检出 307 个致病变异,其中 128 个为新发现变异(图2)。变异类型涵盖错义变异、移码插入变异、移码缺失变异、无义变异(终止获得)、剪接供体 / 受体位点变异、内含子变异、框内插入变异、框内缺失变异、重复扩增变异、同义变异以及 CNV。错义变异占比最高(142/307),其次为终止获得变异(42/307)与移码缺失变异(41/307)。
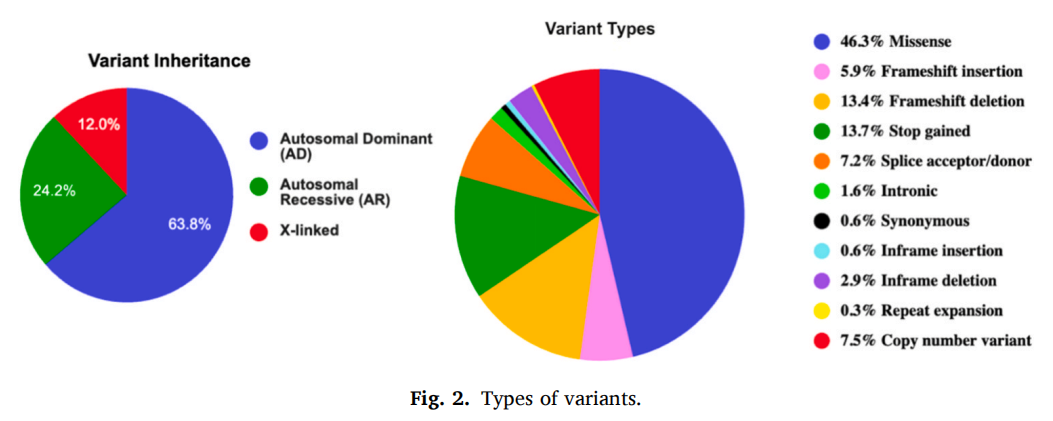
图2
260 例确诊患者累及全身多系统脏器,临床表现多样(表 1);神经系统异常为该队列最常见首发症状,其次为头颈部畸形与肌肉骨骼系统病变。

表1
9 例患者检出双重分子基因学诊断,检出变异对应的基因表型和患者临床症状完全吻合。有5名患者存在拷贝数变异(CNV)。因此如果这5名患者仅进行CMA,就可能遗漏了这些同时存在的单基因疾病。
总计 21 例受试者确诊携带致病性 CNV,CNV 片段跨度 74.6kb~34Mb,变异相关表型与文献报道疾病特征一致。21 例 CNV 阳性病例中仅 1 例为新发 CNV 变异:该患者携带 PKD1 基因 1~9 外显子杂合致病性 78.5kb 缺失,关联 1 型常染色体显性多囊肾病(OMIM#173900);患儿临床表现为智力障碍、特殊面容、孤独症,15 岁出现泌尿系统症状行肾脏超声偶然发现双侧肾囊肿,无家族肾囊肿或肾衰竭病史;因家属失访未完成父母 PKD1 基因验证。
研究检出多种高发遗传病。努南综合征为本队列最高发病种,共 7 例确诊,致病基因分别为 PTPN11、RAF1、RIT1、KRAS、NRAS、CBL、LZTR1;心面皮肤综合征 3 例(BRAF 突变 2 例、MAP2K2 突变 1 例)。7 例努南综合征患者中仅 4 例依靠临床体征疑似诊断(RIT1、NRAS、CBL、LZTR1 相关亚型);马来西亚本地仅可开展 PTPN11 单基因Sanger测序,其余亚型无法通过常规检测完成分子确诊。
1 型神经纤维瘤病(OMIM#162200)共 6 例确诊,其中 1 例患者存在17q11.2 大片段缺失。全部病例均满足神经纤维瘤病临床确诊标准。该大片段缺失患者临床表现为多发牛奶咖啡斑、16 岁起病的多发神经纤维瘤、轻度智力低下、眼距增宽、脊柱侧弯、动脉导管未闭、室间隔缺损。
1 型歌舞伎综合征(KMT2D 致病,OMIM#147920)确诊 5 例,KBG 综合征(ANKRD11 致病,OMIM#148050)确诊 4 例;回顾诊疗记录显示 WES 检测前仅 3 例临床疑似歌舞伎综合征、1 例可疑 KBG 综合征,凸显 WES 在罕见病隐匿病例检出中的重要价值。
已明确遗传分型的 268 例确诊病例遗传方式:常染色体显性遗传 63.8%(171/268)、常染色体隐性遗传 24.3%(65/268)、X 连锁遗传 11.9%(32/268)。受经费限制本研究仅检测先证者,未同步完成父母样本测序,无法区分变异为新发突变或亲代遗传;所有确诊病例父母表型正常,但常染色体显性遗传病轻症携带父母、嵌合体父母、隐性遗传病携带者亲代、X 连锁遗传病携带母亲大多无明显临床症状,依靠体格检查无法排除。
82%(401/489)受试者自愿接收测序次要分析;其中 5.5%(22/401)检出心血管或肿瘤相关基因 P/LP 变异(心血管相关占 64%,14/22;肿瘤相关 27%,6/22)。MYH7、MUTYH、LDLR、TNNT2 基因的致病性次要变异各检出 2 例。22 例次要发现阳性病例中仅 2 个家庭完成家系逐级筛查,其余家庭因经济困难无法开展家系验证;所有家属收到次要发现报告后均无明显不良情绪。
当WES未检出与送检主适应症相关的致病结果时,出具次要分析结果尤为棘手;患者及其家属往往难以接受该结果,进而加重心理焦虑。本研究中 22 例受试者里有 13 例未能明确自身原发疾病病因,在此前提下检出次要变异结果,可能进一步加剧家属的挫败感与心理冲击。上述情况凸显了次要发现带来的经济、心理层面潜在影响,需在全外显子测序术前知情告知环节充分沟通说明,帮助受试家庭做好心理建设,从容接纳各类检测结局。
病例 1:早期分子确诊的临床获益
5 月龄男婴,足月顺产,父母非近亲婚配,为家中首胎(图3,家系1);出生阿氏评分偏低,出生后呼吸机辅助通气 34h、抗生素抗感染治疗,未采用亚低温疗法。生后第 7 天精神萎靡、反复惊厥发作,脑电图提示右侧中央区间断癫痫样放电,颅脑 MRI 未见异常;予抗癫痫药物(ASM)治疗,3 月龄停药后惊厥完全控制;5 月龄查体仅见小头畸形,神经系统查体无其他异常。患儿 9 月龄因小头畸形合并轻度发育迟缓完善 WES 检测,检出 NTRK1 基因 1 个致病变异、1 个可能致病变异:NM_002529.4:c.1196–3_1196–1del p.(?) 与 c.2046dup p.(Val683CysfsTer21);上述复合变异既往已被证实关联 NTRK1 相关先天性无痛无汗症(NTRK1-CIPA,OMIM#256800);家系分型证实两个变异呈反式排列。患儿 1 岁出现特征性自残表现:反复啃咬口唇、舌头、手足,四肢、口腔黏膜多发溃疡瘢痕;临床最终确诊 NTRK1-CIPA,随访合并角膜溃疡、全面发育迟缓、多动。
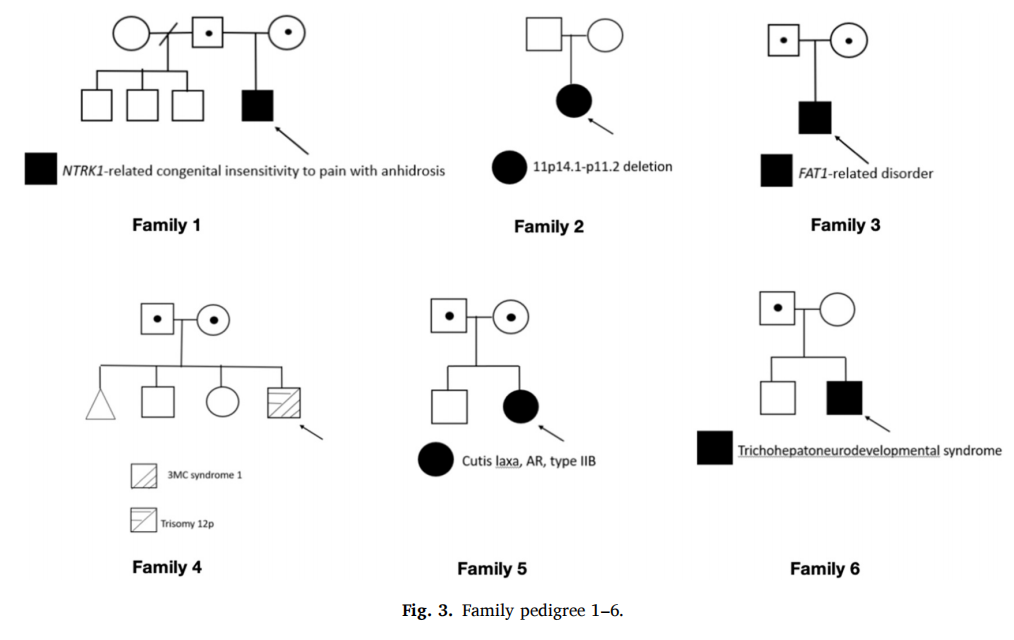
图3
NTRK1-CIPA 为常染色体隐性遗传病,典型特征为痛觉缺失、无汗、智力发育异常,致病原因为 NTRK1 基因双等位功能缺失突变。日本、以色列贝都因人族群存在特有热点突变:日本患者中 p.Phe284TrpfsTer36、p.Arg554GlyfsTer104、p.Asp674Tyr 三个变异占致病突变总数 70%;汉族人群高频突变为 p.Phe284TrpfsTer36;以色列贝都因人族群 p.Pro621SerfsTer21 突变占全部致病突变 89%。
Shalimar等人此前已在马来西亚家系中报道过NTRK1-CIPA病例,3 名患病患儿携带NTRK1基因复合杂合突变:V709L 与 G718S 突变。本研究中这名马来裔患儿同样检出NTRK1复合杂合变异,变异位点为 NM_002529.4:c.2046dup,p.(Val683CysfsTer21) 以及 c.1196–3_1196–1del,p.(?)。值得关注的是,本中心另 1 例非本组队列的马来裔患者检出上述第二种变异的纯合基因型;该患儿 9 月龄起出现自残行为伴全面发育迟缓。患儿除反复发热外,还合并多种骨科并发症:左侧肘关节反复化脓性关节炎、复发性骨髓炎、双侧髋关节脱位、右股骨骨折不愈合及手指自发性离断,拔除牙齿后自残症状有所缓解。
该患儿在自残症状出现前就已提前获得基因确诊结果。早期诊断结合预见性健康指导,帮助家属与临床医护充分认识病情、优化诊疗方案。为缓解家属在疾病初期的心理焦虑,临床对接了同病种患病家庭,为患儿家庭提供社会心理帮扶。
病例 2:早期基因确诊对长期随访监测的重要意义
新生儿重症监护室(NICU)转诊女婴,特殊面容,父母非近亲婚配,首胎(图3,家庭2);孕 28 周检出宫内生长受限,足月出生体重 1.88kg(小于同胎龄);合并先天性甲减、巨大室间隔缺损致心力衰竭,反复肺炎败血症需长期呼吸机支持,生长落后,伴低位耳、睑裂下斜、鼻梁扁平、小下颌畸形。患儿 2 月龄 WES 检出 11p11.2p14.1 区段 17Mb 杂合缺失,缺失片段同时覆盖 WAGR 综合征(肾母细胞瘤、无虹膜、泌尿生殖畸形、智力障碍,OMIM#194072)与Potocki-Shaffer连续基因缺失综合征(OMIM#601224)致病区间。父母染色体核型均正常。
依托基因诊断结果早期转诊眼科,确诊双眼无虹膜、白内障、上睑下垂;婴儿期肾脏初筛超声无异常,但 8 月龄筛查确诊双侧 Ⅲ 期肾母细胞瘤,行双肾保留手术联合放化疗;随访至 2 岁 7 月龄,患儿矮小、小头畸形、轻度全面发育迟缓、右耳听力受损,无多发性外生骨疣。本例说明了早期进行基因诊断对于后续监测和治疗的重要性。
WAGR综合征是一种罕见的微缺失综合征,涉及部分或全部染色体带11p13。PAX6、WT1和其他基因的半合子性是该综合征的主要特征。Potocki-Shaffer综合征由 11p11-p12 片段缺失导致。在该综合征中,EXT2 缺失引发多发外生骨疣、ALX4 缺失致顶骨孔扩大。其他描述的Potocki-Shaffer综合征的表型包括畸形、发育迟缓、眼科异常和男性生殖器异常。
据现有文献检索,本例为同时合并 WAGR 综合征与Potocki Shaffer综合征片段缺失的第 4 例报道病例。1995 年McGaughran等人首次报道 1 例 26 岁男性 WAGR 综合征患者,伴多发性骨软骨瘤:患儿 2 月龄确诊双眼无虹膜、左顶骨大面积颅骨缺损,2 岁罹患肾母细胞瘤,合并智力障碍;部分骨软骨瘤需外科手术切除。染色体核型检出 11 号染色体 11p11.2-p14.2 区间中间缺失,提示该区域存在多发性骨软骨瘤致病候选位点。
2005 年Dominique报道第 2 例经核型与 FISH 确诊的复合型缺失病例:一名 25 岁女性,3 月龄发现无虹膜,15 月龄确诊肾脏肿瘤,6 岁骨骼筛查检出长骨多发骨软骨瘤;伴中轻度智力发育迟缓、行为异常,需抗精神病药物干预。近期另有 1 例 5 月龄女婴被报道罹患该 WAGR和Potocki Shaffer连续片段缺失综合征,临床表现为无虹膜、前囟膨隆及发育迟缓,经CMA证实染色体缺失。
病例 3:测序数据再分析的临床价值
一名男性患儿因智力障碍、双侧先天性上睑下垂、睑裂狭小就诊,初次全外显子测序(WES)结果阴性(图 3,家系 3)。间隔 1 年后重新分析,在新收录致病基因FAT1中检出两处未被报道的疑似致病性无义变异:NM_005245.4:c.10590C>G,p.(Tyr3530Ter) 与 c.4552G>T,p.(Glu1518Ter);家系分离验证证实两处变异呈反式排列。结合基因型 - 表型关联分析,确诊为FAT1相关遗传病。
该病特征性眼部表现包含上睑下垂、眼缺损、小眼畸形,肾脏受累程度个体差异极大,可完全无肾脏异常,也可出现蛋白尿甚至终末期肾病。FAT1基因型与临床表型存在一定关联:错义突变多诱发激素耐药型肾病综合征,肾外表现多样化;截短变异则以特征性上睑下垂(可伴或不伴眼缺损)、足部并趾为核心表现,肾脏病变轻重不一,从无症状、微量蛋白尿、肾病综合征进展至需肾移植的终末期肾病均有报道。本例患儿携带截短变异,除上睑下垂、睑裂狭小外,仅存在无症状镜下血尿与蛋白尿,肾功能指标正常,已转诊肾病专科随访。该病例印证:即便初次测序阴性,持续复盘重分析对明确病因至关重要。
病例 4:CNV检测的重要意义
16 月龄男童,全面发育迟缓、全身性肌张力低下、颅缝早闭伴前额高耸、传导性耳聋、眼距增宽、睑裂狭小、双侧眼球震颤(图 3,家系 4)。先证者初筛 WES 结合父母家系验证,在MASP1基因检出复合杂合VUS,与常染色体隐性 3MC 综合征 1 型(又称颅缝早闭伴眼睑畸形,OMIM# 257920)表型匹配。3MC1 型为罕见常染色体隐性多发畸形病,典型表现:眼距宽、睑裂狭小、上睑下垂、眉弓高拱、唇腭裂、生长及发育落后、听力损伤,亦可合并颅缝早闭、尺桡骨骨性融合、生殖器发育异常。
时隔两年,依托实验室分析流程与数据库更新再次复盘,检出 12 号染色体 12p13.33-p11.1 区间一处 34Mb 的杂合致病性重复变异。12p 三体患儿存在特征性特殊面容,表型与Pallister-Killian综合征(PKS,OMIM# 601803)有相似之处:面型扁平、面颊饱满、前额突出伴发际线偏高、眼距增宽、睑裂短、鼻短小、鼻背宽平、人中偏长、上唇薄、下唇外翻、下颌突出、低位耳;但相较于 PKS,12p 三体患儿神经系统预后更佳,极少合并重要脏器严重畸形。综上,该患儿为孟德尔常染色体隐性遗传病 + 染色体拷贝数变异病双重确诊。
讨 论
本研究仅对先证者开展WES,入组患者均已完成完善临床评估,但仅做过少量基础基因检测。本队列仅先证者测序的初始确诊率为 50%,与既往不同表型病种队列报道的 25%~58% 确诊率基本持平。本研究确诊率偏高,部分原因在于多数受试者将 WES 作为一线基因检测,此前未接受CMA、单基因检测或基因panel检测。研究者推测,若同步采集父母样本行三人组家系测序(trio-WES),整体确诊率有望进一步提升,已有研究证实三人组测序检出效能更高 。三人组测序有助于精准评估遗传风险、开展遗传咨询,一名 23 岁遗传性皮肤松弛症(CL)女性病例即可佐证(图 3,家系 5):患者表现为皮肤松弛无弹性、早老样貌、关节过度伸展,无皮肤外脏器受累;WES 检出PYCR1基因两处新发疑似致病性复合杂合变异,分别源自父母双方,最终确诊常染色体隐性 ⅡB 型皮肤松弛症(OMIM#612940)。遗传性皮肤松弛症遗传模式多样,包含常染色体显性、常染色体隐性、X 连锁隐性,脏器并发症严重程度直接决定预后。该病临床表型易初步怀疑,但明确遗传分型、预判内脏受累必须依靠分子诊断,同时也能为此类育龄女性提供关键的生育遗传指导。
尽管三人组 WES 可凭借明确的变异连锁相位信息获得更高确诊率,但部分病例仍需后续补充验证,17 岁常染色体隐性毛发 - 肝脏 - 神经发育综合征(THNS,OMIM#618268)男性患者即为典型(图 3,家系 6):患儿中度智力障碍、脊柱侧弯,既往在儿科按痉挛型脑瘫随访。CCDC47基因一处疑似致病性杂合无义变异 NM_020198.3:c.1211_1232dup,p.(Arg412LysfsTer4) 遗传自父亲;而另一杂合意义未明变异 c.669+5G>T,p.(?) 的剪接 AI 评分为 0.65,在患儿母亲外周血与口腔黏膜样本中均未检出,判定为新发变异。该分子诊断可完整解释患儿羊毛状发、特殊粗陋面容、一字眉、厚唇、远端关节挛缩、脊柱侧弯及全面发育迟缓的临床表现,患儿无肝功能异常。后续仍需进一步试验,明确该新发变异源自父方或母方染色体。
THNS 于 2018 年由Morimoto团队首次报道,4 例患者均为多系统受累,典型特征包括羊毛状发、肝功能损伤、皮肤瘙痒、特殊面容、肌张力低下及重度发育迟缓 ;近年国内报道 1 例中国患儿,以新发听力受损为特征性并发症。已报道病例的CCDC47致病变异均为无义变异或移码变异。据现有文献,本例为本病第 6 例报道病例,也是目前确诊年龄最大的患者。
WES 联合拷贝数变异测序(CNVseq)可显著提升确诊率(P=0.016)。本研究共 21 例经 WES 检出拷贝数变异,其中 5 例通过其他方法完成验证:2 例(5 号、8 号患者)采用 FISH 验证 CNV,2 例(9 号、19 号患者)行 CMA 检测。患者14的Angelman综合征通过Prader Willi综合征/Angelman综合征的甲基化特异性-多重连接依赖探针扩增被证实。马来西亚当地 CMA 检测资源有限、普及度不足,WES 虽无法 100% 精准检出 CNV,但兼具变异筛查与拷贝数分析能力,是资源受限地区优选检测方案。
全队列共 9 例(1.8%)患者得到双重基因诊断,仅依靠临床查体很难实现该类复合疾病确诊。Posey团队在 7374 例外显子测序中检出 1.4% 的双重遗传病,与本研究数据接近。若患者临床症状无法用已确诊的单种遗传病完全解释,需警惕合并第二种遗传性疾病。相较于定向基因panel检测,WES 无偏倚覆盖全部编码区基因,无需临床医生提前甄别拆分复杂表型,是排查复合遗传病的优选方案。
本研究共 16 例确诊 RAS 通路病。RAS 通路病是一类表型高度重叠的遗传病总称,致病根源为 RAS/MAPK 通路相关基因的胚系致病性变异,颅面部特征与努南综合征高度相似,而努南综合征是本组疾病中最常见亚型。努南综合征最主要致病基因为PTPN11(占 50%),其次为SOS1(16%~20%);其余 RAS 通路病包括科斯特洛综合征、心 - 面 - 皮综合征、多发性雀斑样痣努南综合征及 1 型神经纤维瘤病。本研究 16 例 RAS 通路病中 7 例为努南综合征,致病基因分散,未检出SOS1突变。
除本研究检出的新发变异外,也检出已知致病变异,两例马来裔患儿可举例佐证:两名患儿新生儿期即出现溶血性贫血,需规律输血,WES 确诊 伴溶血性贫血的4 型远端肾小管酸中毒(RTA)(OMIM#611590)。二人SLC4A1基因均携带已知疑似致病性变异 NM_000333.1:c.1199_1225del,p.(Ala400_Ala408del),分别搭配两种不同新发意义未明复合杂合变异(c.2002G>A,p.(Ala668Thr)、c.1319T>A,p.(Leu440Gln))。已知SLC4A1p.Ala400_Ala408del突变可导致东南亚卵圆细胞贫血(SAO)(OMIM#166900)。SAO通常无症状。然而,SLC4A1基因中的双等位基因突变与常染色体隐性型远端肾小管酸中毒4相关,并伴有溶血性贫血,这是热带地区(尤其是马来西亚、泰国、菲律宾和巴布亚新几内亚)远端RTA的常见病因。在非热带国家,SLC4A1突变引起的远端RTA是罕见的,几乎总是以常染色体显性方式遗传。隐性SLC4A1突变导致的远端RTA通常出现在儿童早期,而显性SLC4A1突变导致的远端RTA发病较晚。
Narazah等人在马来西亚东海岸吉兰丹州(马来裔占 95%)开展流行病学研究,当地 82% 远端RTA患者合并 SAO,与本研究两例病例特征一致。SAO在马来族群中的高流行率(约4%)可能是由于自然选择对抗疟疾感染的结果,因为吉兰丹州仍存在多个农村地区,疟原虫感染率较高。鉴于该国SAO的高流行率,已有报告指出存在纯合型SAO患者。纯合型SAO患者在产前或出生时即被检测出贫血,并伴有胎儿水肿、肝脾肿大等症状。这些患者需在宫内输血、产后输血以及出生后接受重症监护治疗。多数患者在新生儿期或婴儿期死亡。
入组患者中 401 例(82%)自愿接收次要发现结果,提示受试者普遍愿意获取和自身健康管理相关的附加基因信息,与既往研究结论一致。依据美国医学遗传学与基因组学学会(ACMG)指南推荐的 59 种临床可干预基因,本研究 22 例(5.5%)检出致病性 / 疑似致病性次要变异,和全球外显子 / 全基因组研究 1%~5% 的次要变异检出比例相符。报告此类可干预变异有助于生育规划、家系高危个体级联筛查、针对性临床随访,规避不必要检查,但相关远期获益仍缺少大样本循证证据;现有研究证实披露次要发现不会对受试者造成显著负面心理影响,也不会降低患者接受基因检测的意愿。
1 例无肿瘤家族史的马来裔患者,次要发现检出BRCA2杂合致病变异 c.262_263del (p.Leu88AlafsTer12)。BRCA2为抑癌基因,突变显著升高乳腺、卵巢、前列腺及胰腺癌发病风险(OMIM#612555)。该变异 2008 年由Thirthagiri首次在马来西亚两名无亲缘乳腺癌马来患者中报道,其团队筛查 187 例当地乳腺癌患者后发现马来裔乳腺癌致病突变以BRCA2变异为主;2017 年Wei Xiong等纳入 2000 余例马来西亚乳腺癌病例与 2000 余例健康对照,证实马来裔乳腺癌患者中 44%(7/16)BRCA2突变为 c.262_263del,健康对照组该变异占全部BRCA2变异的 50%。
另有两名二十岁成年患者携带相近LDLR杂合疑似致病变异 c.1721G>A (p.Arg574His),分别为马来裔女性、华裔男性。LDLR突变致常染色体显性家族性高胆固醇血症(FH,OMIM#143890),以血浆低密度脂蛋白胆固醇显著升高为核心表现,携带者早发冠心病风险大幅上升。全球人群杂合 FH 发病率 1/500,纯合型约 1/100 万,患者血脂、黄瘤、心血管病变表型个体差异明显;杂合男性近 80% 罹患冠心病,女性仅 20%~30% 发病。本例两名携带者血脂指标完全正常,检出变异后可启动家系级联筛查与长期随访,及早防控早发冠脉病变。马来西亚既往多项 FH 突变谱研究未在本土人群报道LDLR c.1721G>A 变异,仅在中国台湾汉族患者中有文献记载。
本研究整体分子确诊率 53%。针对初筛阴性病例,完善患者及家系深度表型采集,可指导甲基化检测等 WES 常规无法覆盖的补充试验。定期数据重分析可提升 10%~15% 确诊率,埃尔多梅里预实验中重分析甚至将确诊率提升至 51%;新增致病基因收录、补充三人组测序、新发变异与 CNV 分析算法迭代是重分析提升检出率的核心因素。临床与检验科室及时同步新增临床症状、疑似诊断,可进一步优化重分析成效。
阴性病例另一备选方案为全基因组测序(WGS)Belkadi等人的研究证实 WGS 在单核苷酸变异、小插入缺失检出效能上优于 WES,测序深度均一性、分型准确度更高、假阳性更低;大片段结构变异、深度内含子变异是大量罕见病的致病根源,Pagnamenta等对 122 例罕见病患行 WGS 后发现,结构变异、剪接位点变异、深度内含子变异贡献了 43% 疑难病例的病因确诊。
研究局限性:53% 偏高的确诊率源于本中心 CMA、单基因及基因包检测资源匮乏,多数患者直接选用 WES 作为首筛;入组病例 83% 为儿童,结论无法代表晚发型遗传病人群特征;受项目经费限制未能常规采集父母样本,无法开展三人组测序,既阻碍隐性遗传病变异相位解析,也难以明确变异来源。
本研究是马来西亚首个大样本 WES 临床研究,单中心 489 例罕见病患者中 260 例(53%)通过分子测序明确致病基因。结果证实 WES 在马来西亚罕见病临床诊断具备实用价值,可缩短患者漫长的就医确诊周期,实现精准分子分型,支撑靶向治疗、个体化随访与遗传咨询。联合国 2019 年卫生政策宣言已将罕见病纳入全民健康保障建设范畴,在马来西亚落实 WES 检测经费保障,是落地罕见病关怀相关联合国决议的重要举措之一。
参考文献:
Moey L H, Seo G H, Cheah B E, et al. A first large study of whole-exome sequencing (WES) in 489 patients with suspected rare genetic disorders at a tertiary centre in Malaysia[J]. Rare, 2025, 3:100102. https://doi.org/10.1016/j.rare.2025.100102
小提示:本篇资讯仅在梅斯医学APP中开放阅读,请扫描二维码直接下载APP
