深度解析医学证据,DeepEvidence为你支撑决策
2026年5月23日,中山大学附属第六医院邓艳红教授团队在 Gut 期刊发表研究论文,首次揭示了MSS结直肠癌中PTEN缺失导致免疫逃逸的新机制。研究发现,PTEN缺失通过KEAP1-NRF2-自噬轴促进MHC-I降解,使肿瘤逃避CD8⁺ T细胞杀伤。研究同时提出,靶向NRF2联合抗PD-1治疗可有效逆转该耐药表型,为MSS肠癌患者提供了新的治疗策略。

研究背景
免疫检查点阻断(ICB)治疗,尤其是PD-1/PD-L1抗体,在MSI-H(微卫星高度不稳定)型结直肠癌中取得了显著疗效。然而,MSI-H仅占全部结直肠癌的15%左右,剩下约85%的MSS(微卫星稳定)型患者对免疫治疗普遍不敏感。
这种“冷”肿瘤微环境是MSS型结直肠癌免疫治疗耐药的核心原因,具体表现为:肿瘤浸润CD8⁺ T细胞数量少、肿瘤突变负荷低、抗原加工和呈递能力缺陷。
PTEN是一个经典的抑癌基因,其脂质磷酸酶活性介导的PI3K-AKT信号通路抑制是其主要作用机制。在结直肠癌中,PTEN基因突变发生率约为8%-10%,但PTEN蛋白表达缺失的比例高达19%-36%,尤其在MSS亚型中更为常见。这种蛋白水平的缺失不仅源于基因突变,还与表观遗传沉默和翻译后调控有关。然而,PTEN蛋白缺失是否直接导致MSS结直肠癌的免疫逃逸和ICB耐药,此前尚不明确。
主要发现
1. PTEN缺失与免疫逃逸密切相关
临床数据显示:
● PTEN低表达的患者接受PD-1治疗后,进展比例更高,生存期更短。
●PTEN低表达的肿瘤中,CD8⁺ T细胞浸润显著减少,且T细胞功能耗竭(GZMB、IFN-γ下降)。
●单细胞测序显示,PTEN缺失肿瘤中的CD8⁺ T细胞向终末耗竭状态的分化受阻,细胞毒性分子表达下调。
2. PTEN通过非经典通路调控免疫(不依赖AKT)
传统认为PTEN主要通过抑制PI3K-AKT通路发挥作用,但本研究发现AKT抑制剂无法逆转PTEN缺失带来的免疫逃逸表型,提示存在其他机制。
3. 机制:PTEN稳定KEAP1,抑制NRF2
● PTEN通过C2结构域与KEAP1的DGR结构域直接结合。
● 这种结合阻止了p62对KEAP1的自噬性降解。
● PTEN缺失 → KEAP1降解 → NRF2释放并过度激活。
4. NRF2过度激活导致MHC-I降解
过度活化的NRF2:
● 转录上调自噬受体NBR1及自噬相关基因(ULK1、ATG5等)。
● 驱动MHC-I进入自噬-溶酶体途径降解。
● 导致肿瘤细胞表面MHC-I显著下降。
MHC-I是CD8⁺ T细胞识别肿瘤的关键分子,其丢失使T细胞无法有效杀伤肿瘤。
5. NRF2同时抵抗铁死亡
PTEN缺失的肿瘤细胞还表现出抗铁死亡基因(GPX4等)上调,对铁死亡诱导剂不敏感,进一步增强肿瘤生存能力。
治疗策略:NRF2抑制剂联合PD-1抗体
1. ML385恢复MHC-I表达
使用NRF2抑制剂ML385处理PTEN缺失的肿瘤细胞:
● MHC-I总蛋白和表面表达水平显著恢复。
● MHC-I在溶酶体和自噬体中的积聚减少,重新分布至细胞膜。
2. 联合治疗在动物模型中效果显著
在多种小鼠模型中(皮下移植瘤、直肠原位瘤、AOM/DSS诱导的自发肠癌模型),ML385与抗PD-1抗体联合治疗显示出协同效应:
● 肿瘤生长被显著抑制,肿瘤负荷减轻。
● 小鼠总生存期延长。
● 肿瘤内CD8⁺ T细胞浸润增加,GZMB⁺、IFN-γ⁺、Ki-67⁺的活化CD8⁺ T细胞比例升高。
● IHC染色证实联合治疗组CD8、GZMB、IFN-γ表达升高,Ki-67降低。
3. 联合治疗增强铁死亡敏感性
ML385与erastin联合使用,相比单药,进一步降低PTEN缺失细胞的活力,增加脂质过氧化和活性氧积累。
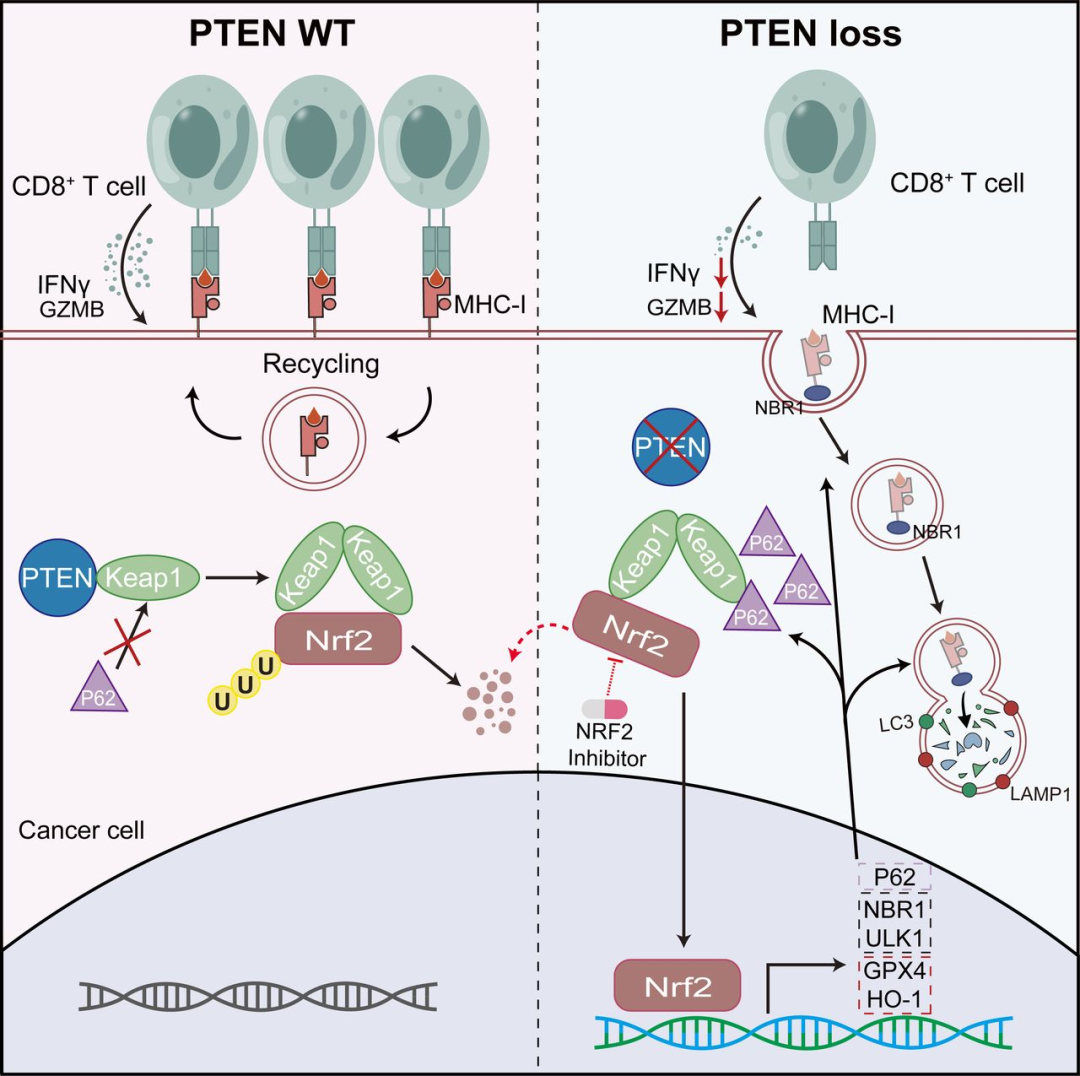
这项研究首次揭示:PTEN通过其C2结构域与KEAP1结合,竞争性抑制p62介导的KEAP1降解,从而维持NRF2的基础水平。PTEN缺失打破这一平衡,导致NRF2过度激活,进而通过自噬途径降解MHC-I,使肿瘤逃避CD8⁺ T细胞的识别和杀伤。靶向NRF2的药物ML385能够同时恢复抗原呈递和铁死亡敏感性,并协同增强抗PD-1治疗的疗效。
参考资料:Cai R, Zhan W, Lyu X, et al. PTEN deficiency impairs MHC-I-mediated tumour immunity via NRF2-dependent autophagy in microsatellite stable colorectal cancer. Gut. Published online May 22, 2026. doi:10.1136/gutjnl-2026-338195