首页 > 疾病防控/ 正文
深度解析医学证据,lxfs.net为你支撑决策
供者细胞源性血液肿瘤(DDHN)是一类罕见疾病,目前缺乏标准化的诊断标准和治疗管理方案。基于此,来自欧洲、美洲和澳大利亚的异基因移植及血液恶性肿瘤领域国际专家(代表EBMT实践协调与指南委员会)共同制定了DDHN的实用性诊断框架,并就后续临床管理发布指导意见。专家组于2025年9月18-19日在德国柏林召开会议,在深入文献复习的基础上,讨论了DDHN的定义、分子机制、治疗策略及供者结局。
相关建议近日发表于《Lancet Haematology》,介绍了DDHN的流行病学和临床定义,提供了诊断和预防指导,阐述了治疗考量,并概述了供者管理的推荐意见。

引言
异基因造血细胞移植(HCT)是许多血液恶性肿瘤(尤其是高危急性白血病和慢性髓系肿瘤)患者的唯一根治性手段,移植失败的主要原因为疾病复发(发生率为10-50%,取决于疾病风险)以及移植相关并发症。在少数情况下,移植后出现的血液肿瘤并非来源于原发克隆或其演化,而是源自供者造血细胞。
50多年前,首例供者细胞源性急性淋巴细胞白血病(ALL)被报道,此后有多种类型的DDHN被陆续报道,最常见的是急性髓系白血病(AML)、骨髓增生异常综合征(MDS)或骨髓增殖性肿瘤(MPN);淋巴增殖性疾病亦有偶发报道。DDHN报告频率的增加可能由多种因素导致,包括异基因HCT活动量增加、移植受者总生存期延长,以及对DDHN认知和识别能力的提高——尤其是分子工具(如灵敏的嵌合体分析和靶向基因测序)的发展,使得检测供者细胞来源的分子异常成为可能。尽管当前供者筛查政策使得显性白血病克隆的传播不太可能发生,但克隆性造血在健康人群中是公认现象,在多达四分之一的造血干细胞供者中可被检测到。此外,当前分子平台的应用揭示了相当数量DDHN患者存在体质性遗传变异(如DDX41),尤其是在亲属供者中,可对其进行体质性疾病筛查。
重要的是,一部分供者在其受者发生DDHN后的动态随访中,自身也出现了血液恶性肿瘤,且其临床和分子特征常与受者的DDHN相似。因此,DDHN的诊断涉及供者告知、纵向监测及后续临床管理等方面的重要伦理考量。
关于DDHN发生的危险因素,现有研究结果存在争议且异质性较大。DDHN的总体预后一致报告为不良,即使经过治疗也是如此。
鉴于DDHN在诊断和管理上的挑战,坐着组建了国际专家小组,以制定实用性诊断框架并指导后续临床管理。需注意,移植后淋巴增殖性疾病(PTLD)因其具有独特的、常由病原体驱动的病理生理机制,不在本讨论范围内。
定义与流行病学
DDHN通常定义为源自移植供者造血干细胞的新发(de-novo)血液恶性肿瘤,与原受者的肿瘤无关。DDHN最常表现为髓系疾病(如AML、MDS、MPN或MDS/MPN),但淋巴系肿瘤也可发生。
由于定义、检测方法和报告程度的异质性,精确的发病率估算受到限制。此外,多数研究基于旧有队列,缺乏当前分子知识和方法,可能导致DDHN频率被低估。EBMT登记组数据显示较为DDHN罕见,异基因HCT后10年累计发生率为0.132%。在该研究报告的38例DDHN中,22例(58%)为AML,7例(18%)为MDS,2例(5%)为ALL,2例(5%)为慢性髓系白血病(CML),5例(13%)为慢性淋巴细胞白血病(CLL)。从移植到DDHN诊断的中位时间为44个月(范围2-279个月)。在一项日本登记组研究中,DDHN的10年发生率估计为0.13(SD 0.02)。该研究描述的40例DDHN中,10例(25%)为供者源性AML,23例(58%)为供者源性MDS,中位诊断时间为22个月(范围3-190个月)。在最近一篇回顾2009-2024年间95例DDHN的综述中,54例(61.5%)为AML,20例(23%)为MDS,7例(8%)为ALL,2例(2%)为CML,5例(5.5%)为其他疾病(1例为慢性粒单核细胞白血病,1例为多发性骨髓瘤,1例为滤泡性淋巴瘤,1例为慢性淋巴细胞白血病)。。
DDHN可发生于任何干细胞来源的异基因HCT后。供者与受者的亲缘关系和干细胞来源的影响尚不明确,存在矛盾数据。例如,有报道称DDHN更常见于无关脐血移植后,而亲缘或无关供者移植后均有发生,无明显倾向。在最新的EBMT报告中,与配对的对照病例相比,受者使用粒细胞集落刺激因子(G-CSF)和体内T细胞耗竭与DDHN显著相关(χ²p<0.05)。前述日本登记组研究中,脐血移植和供者年龄较大是危险因素,但使用亲缘供者未增加DDHN风险(即使排除脐血)。然而该研究开展于当前大多数体质性基因突变被系统性筛查之前,如今来自亲缘供者的DDHN发生率可能更高,有待未来研究验证。
DDHN预后较差,长期存活者少,中位总生存期因DDHN类型和研究而异。在最新的EBMT研究中,从DDHN到死亡的中位时间为11个月(范围0-91个月)。在Tsirigotis等人的综述中,有数据的75例患者中45例死亡,中位总生存期估计为36个月。
由于DDHN可能与导致移植的原发疾病属于同一细胞系,其发病率可能被低估——部分病例可能被误认为原发病复发。同样,登记处数据库中也存在漏报可能。因此,随着对DDHN认识的提高和能明确判定供者来源的检测方法的广泛应用,其观察到的发病率可能会增加。
分子机制
DDHN涵盖异质性疾病谱,可能呈现不同的发病途径(图1)。曾有报道称,供者的完整白血病克隆在不知情的情况下被转移(供者通常在捐献后不久即被确诊),但鉴于当前的供者筛查措施,该情况极为罕见。
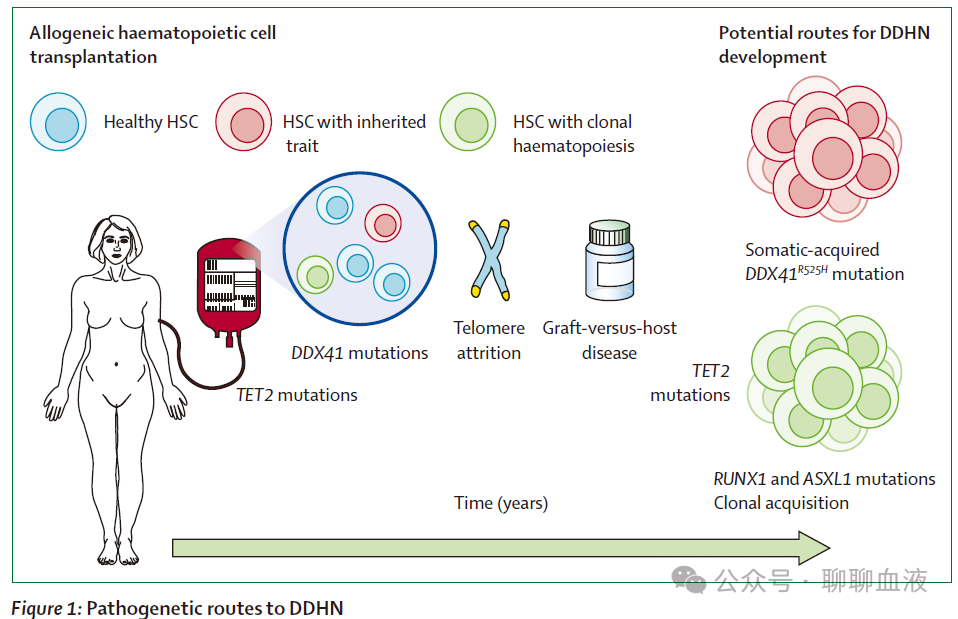
与之相反,许多DDHN病例与携带体质性突变的干细胞转移有关,这些突变已知与髓系恶性肿瘤的易感性相关(例如CEBPA、DDX41、GATA2、RUNX1、XPD、XRCC1、CHEK2和TERT)。DDHN诊断后,这些突变常在相应供者中得到确认。在胚系突变中,DDX41是目前DDHN患者中最常见的复发性受累基因,这可能是由于致病性 DDX41 突变携带者缺乏临床症状以及 DDX41 突变在髓系疾病患者中的相对频率。CEBPA 的胚系突变(另一种无先存临床表型的疾病)报道为 DDHN 病因的频率较低,可能是由于其在 2004 年发现了其临床相关性,并且 CEBPA 被广泛纳入临床诊断时的二代测序(NGS)panel 中。干细胞供者的胚系易感状态目前似乎是大多数髓系 DDHN 的原因。
然而部分 DDHN 不具有典型的易感突变,可能是由于缺乏对遗传性特征的筛查或所用方法的局限性。供者中的克隆性造血是 DDHN 发展的另一个潜在初始触发因素,克隆性造血定义为在无明显恶性肿瘤个体的血液中检测到驱动血液肿瘤的基因中存在复发性体细胞突变。在无血细胞减少的情况下,当克隆突变以 2% 或更高的变异等位基因频率存在时,一旦遵循适当的方法学标准,则WHO 将其归类为潜能未定的克隆性造血(CHIP)。CHIP 的患病率随年龄急剧增加:年龄在 55 岁或以上的健康亲缘供者中约 16% 存在 CHIP,最常见于 DNMT3A、TET2 或 ASXL1。研究表明,与髓系恶性肿瘤患者相关的供者相比,与淋系恶性肿瘤患者相关的供者中 CHIP 更常见(19.2% 对 6.3%;p≤0.001),这可能反映了潜在的遗传性易感性,并产生髓系相关的克隆性造血。髓系疾病受者中 CHIP 频率较高可能反映了产生髓系相关克隆性造血的潜在遗传易感性;然而该假说仍需进一步研究。在一篇开创性论文中,对 1727 名年龄在 40 岁或以上的供者样本进行靶向纠错测序,发现 388 名(22.5%)供者存在变异等位基因频率 ≥0.005 的克隆性造血,其中 DNMT3A(253 例 [14.6%])和 TET2(89 例 [5.2%])突变最为常见;克隆性造血存在于 12.6% 的 40-49 岁供者、26.6% 的 50-59 岁供者和 41.2% 的 60 岁或以上供者中。此外,102 个供者克隆中有 86 个(84%)在移植后在受者中显示出长期植入,包括变异等位基因分数低于 0.01 的克隆。然而,从供者克隆性造血进展为 DDHN 需要克隆在可变潜伏期内扩增并获得新的分子事件,或携带未检测到的遗传性特征(在上述研究中的六个测序的 DDHN 病例中有两个)。例如,TET2 突变克隆与较长的潜伏期相关,而 DNMT3A 突变克隆潜伏期较短,因为它们在异基因 HCT 后典型的自我更新和慢性炎症应激条件下扩增更快。供者造血干细胞动员受损,或受者骨髓微环境既往的医源性毒性(例如,既往治疗),都被认为有利于白血病前期细胞的扩增或增殖,类似于细胞毒性治疗后髓系肿瘤发生的原因。研究表明,CHIP(例如TET2)或遗传性特征携带者(端粒基因机制)可影响干细胞动员,因此为未来 DDHN 的发生发展产生潜力。
移植后免疫抑制治疗导致的 T 细胞监视受损也可能促进 DDHN 的发生发展。此外,异基因 HCT 后造血细胞的高复制应激可能在造血重建过程中促进端粒功能障碍和基因组不稳定。尽管脐带血中可能实际上不存在 CHIP,但从较少的干细胞重建骨髓可能会产生类似于再生障碍性贫血患者的瓶颈效应,导致可能获得适应性不良突变,以复杂且尚未完全阐明的方式塑造克隆动态。事实上,7 号染色体改变(治疗相关和再生障碍性贫血后髓系肿瘤的特征)在髓系 DDHN 中很常见(约 25%),暗示残留的受者造血干细胞的内在脆弱性或既往治疗的旁观者效应,或两者兼有,对进入的供者造血干细胞产生影响。
尽管供者克隆性造血显然是 DDHN 的原因,但尚未报道其会降低异基因 HCT 后的总生存期或无病生存期。事实上,供者来源的 CHIP 可降低复发率和减少慢性移植物抗宿主病风险,但由于其回顾性以及大多为单变量分析,这些结果应谨慎对待。
治疗
指导 DDHN 管理的证据很少,大多数数据来自病例报告和系列。治疗策略通常遵循de-novo血液肿瘤的原则。过继性免疫治疗(供者淋巴细胞输注)很少用于治疗 DDHN,因为预期的移植物抗 DDHN 效应低,且存在移植物抗宿主病的潜在危害,因此应避免使用。然而大多数疗法并非根治性,主要作为第二次异基因 HCT(来自不同供者)的桥接,移植仍然是唯一的根治性选择,尤其是对于最常见的 DDHN 表型(例如MDS和 AML)。此外,异基因 HCT 后需要特别注意,因为患者免疫功能低下,且强化治疗导致器官毒性的风险增加。
关于供者来源的 AML,使用强化化疗进行缓解诱导(包括经典的“7+3”AML 诱导化疗或 CPX-351)是重要的基石,尤其是对于移植后晚期发生的白血病(预期毒性低于早期 DDHN)。治疗选择应考虑既往化疗线数、器官储备和累积毒性。在最近一篇 DDHN 综述中,68 名患者中有 35 人在诱导治疗后获得完全缓解。对于因年老或既往癌症治疗而衰弱的 AML 或MDS DDHN 患者,已有报道使用强度较低的方案,特别是去甲基化药物联合或不联合维奈克拉。
全面的分子检测对于识别潜在可靶向的突变并使这种预后不良的情况下能够进行精准治疗至关重要。在 AML 中,针对特定突变的选项包括用于 FLT3 突变 AML 的 FLT3 抑制剂,用于 NPM1 突变和 KMT2A 重排 AML 的 Menin 抑制剂,以及用于 IDH1/2 突变 AML 的 IDH1/2 抑制剂。来那度胺在一例伴有 5 号染色体缺失的供者来源MDS中显示有活性,并且在移植后复发环境中显示出可控的安全性。
关于进行第二次异基因 HCT 后的异基因 HCT 后维持治疗,相关数据很少。一项比较阿扎胞苷与观察的 3 期试验在 AML 或MDS首次异基因 HCT 后维持治疗中结果为阴性。一项 2022 年的荟萃分析报告了阿扎胞苷的优势,但该研究基于回顾性研究和存在许多偏倚的 2 期试验,因此无法推荐这种维持治疗。
第二次移植
尽管第二次异基因 HCT 的应用仅限于病例报告、病例系列和登记处水平的研究,但这种治疗方法在这种高风险环境中已显示出偶发的持久缓解。在当前实践中,由于难治性疾病、体能状态差和合并症、患者拒绝或缺乏合适供者,只有少数 DDHN 患者接受了第二次异基因 HCT。在 Tsirigotis 及其同事的综述中,70 名 DDHN 患者中只有 16 人接受了第二次异基因 HCT,其中两名患者接受了来自同一供者的移植。第二次移植通常使用不同的供者,但也有报告描述了使用同一供者的成功结果。关于第二次异基因 HCT 后的结果,失败的风险因素包括首次异基因 HCT 与复发之间的时间较短、受者年龄较大以及既往有急性或慢性移植物抗宿主病。在 Tsirigotis 及其同事的综述中,接受第二次异基因 HCT 的 16 名患者中有 6 名在中位随访 37 个月后仍然存活。EBMT 最近报道了首次移植后复发时进行第二次异基因 HCT 的令人鼓舞的结果,2 年非复发死亡率为 22%(95% CI 20.6-23.6),复发发生率为 50%(48.1-51.8),总生存率为38%(36.4-39.9),无进展生存率为 28%(26.3-29.7)。有的关于第二次异基因 HCT 的研究重点关注髓系疾病患者,也证实了这些结果。
供者结局
在有数据可用的情况下,受者发生 DDHN 时,供者中也会报告血液恶性肿瘤。在最新的 EBMT 报告中,在受者发生 DDHN 的 25 名供者中,有 7 名也发生了血液恶性肿瘤(其中 5 名供者发生了相同的疾病)。Suárez-González 及其同事描述了文献中的 137 例 DDHN,其中 73 名供者中有 9 名(12%)报告发生了血液恶性肿瘤。William 及其同事估计,在 162 例有供者随访的 DDHN 病例中,有 7 名供者在受者诊断 DDHN 后发生了髓系恶性肿瘤。在 Tsirigotis 及其同事的研究中,在有数据的 44 名供者中,有 7 名在报告时已死亡(无可用数据),有 7 名发生了血液恶性肿瘤,与受者 DDHN 相同,发生在捐献后中位时间 36 个月。
方法学
EBMT 是一个非营利性科学学会,代表主要在欧洲及更远地区的 700 多个移植中心。本综述是在 EBMT 实践协调与指南委员会的方法学指导下产生的。作者检索了 PubMed 数据库中 2000 年 1 月 1 日至 2025 年 6 月 1 日的相关文献,并由移植、髓系疾病、血液病理学和异基因 HCT 领域 4-5 名专家组成的小组进行审查(MR, DHW, AJ, DH, NG, CG, DML, SP, RH, 和 KS)。关键词为“donor-derived leukemia”或“donor-derived malignancy”或“donor clonal hematopoiesis”。专家们确定了关键的未解决问题(panel 1),并在混合研讨会前两个月通过虚拟交流生成了总结性评估,然后在位于德国柏林的为期2天的混合研讨会(2025年9月18-19日)中讨论了这些证据审查和评估。随后生成了一份论文草案(CG、MR、DHW、AJ、DH、RH、MC、JD-S、NAG、JPM、KS和MTV),并分发给来自欧洲、美洲和澳大利亚的同种异体移植和血液恶性肿瘤国际专家(CA、GBa、GBe、EB、YC、FBD、VF、NG、TG、JCH-B、ML、MM、MTM、NP、MQ、CS、FO、KR、AR、IS-O、FC、TC、SP、LP、PS、JV、DPM和IY-A)进行最终修订。由于缺乏随机对照试验的证据,并且该主题的数据通常较少,因此不可能对这些建议进行正式分级,表达的观点是所有作者的专家共识。建议反映了大多数专家意见。当无法达成共识时,作者在建议一节中解释了这些建议的局限性。没有使用外部或行业资金来完成这个项目。
Panel 1:未解决的问题
DDHN 的定义及其流行病学是什么?
DDHN 的预防和诊断指南,包括供者选择,有哪些?
应使用哪些治疗方法(包括细胞治疗)来管理 DDHN 患者?
登记处数据
由于供者来源的白血病是 EBMT 中心收集的一个变量,因此对 EBMT 登记处进行了分析,以全面评估 DDHN 的发生频率。在 2000 年 1 月 1 日至 2023 年 7 月 30 日期间,共识别出 168 例 DDHN 病例,其中 122 例是自 2016 年以来报告的,反映了更全面的分子检测的发展和欧洲异基因 HCT 活动的增加。然而这些数据也证实,与同期登记处记录的 31444 名异基因 HCT 受者相比,DDHN 是一种罕见疾病。需要指出的是,由于报告数据不足以及一些 DDHN 病例被错误归类为原始受者疾病复发,登记处数据可能低估了实际数字。
国际调查
作者就 DDHN 患者的诊断、流行病学和管理发起了一项针对 EBMT 中心的调研(附录第 1-3 页)。需要指出的是,该调研重点关注供者来源的髓系肿瘤,因其是最常见的疾病类型。总体而言,348 个受邀中心中有 104 个(30%)回应了调研(附录第 1-3 页),其中 41 个中心表示至少诊断过一例 DDHN。共报告了 71 例供者来源的髓系肿瘤,其中 47 例来自亲缘供者异基因 HCT。大多数中心表示可以获得扩展的专用 NGS 来表征 DDHN 的分子特征,包括评估胚系易感突变。在 71 例 DDHN 病例中,23 例接受了去甲基化药物治疗,21 例接受了强化化疗,6 例接受了其他治疗,30 例接受了第二次异基因 HCT;10 例未接受治疗。

相关建议
首次异基因 HCT 时的 DDHN 预防
建议对有血液病家族史或临床表现与体质性疾病相符的每位患者或潜在供者进行胚系易感特征筛查(另见 EBMT 最近发布的具体指南)。对符合条件的异基因 HCT 候选者的原始肿瘤(如果是髓系)的分子检查应包括 DDX41。发现胚系易感变异应促使对被视为潜在供者的亲属进行携带者状态评估。如果存在明显的家族性易感性但未识别出明确的体质性突变,建议进行大范围 NGS 筛查,或在可能时进行全基因或全外显子测序。选择哪种 NGS 技术将取决于多个不同因素(例如费用和异基因 HCT 的紧迫性):首选对肿瘤-胚系配对进行全基因或全外显子测序,紧急异基因 HCT可考虑紧急胚系增强体细胞 panel。
在获得更可靠的证据之前,不建议常规筛查供者是否存在克隆性造血或胚系特征。事实上,由于克隆性造血在亲缘和非亲缘供者中的发生率相似,必须考虑这些发现的费用、劳动负担和伦理问题,特别是考虑到其临床可操作性有限。此外,即使在克隆性造血的情况下,DDHN 的发生也很罕见,并且尚未报道供者来源的克隆性造血与异基因 HCT 受者的不良结局相关。
DDHN 的诊断
DDHN 的诊断应满足两个条件:(1)DDHN 细胞为供者来源,尽可能通过高灵敏度方法证明为完全供者嵌合体;(2)有证据表明是新的血液恶性肿瘤,且与首次异基因 HCT 的原始诊断无关。如果 DDHN 与最初导致移植的疾病属于同一疾病类别,则其表型、遗传学或两者可能不同,或可能复制家族中分离的遗传特征(例如,最初在患者中未被筛查的 DDX41 突变,后来在来自亲缘供者的 DDHN 中出现)。
如果可能,可以检测捐献前储存的 DNA 或冻存细胞,以寻找任何与 DDHN 相关的分子异常,这对于胚系易感基因尤为重要。在这种情况下,在 DDHN 和冻存的供者 DNA 中发现共享的分子改变可为白血病的供者细胞起源提供额外证据,尽管这对于 DDHN 诊断并非必需。
DDHN 的常规诊断检查
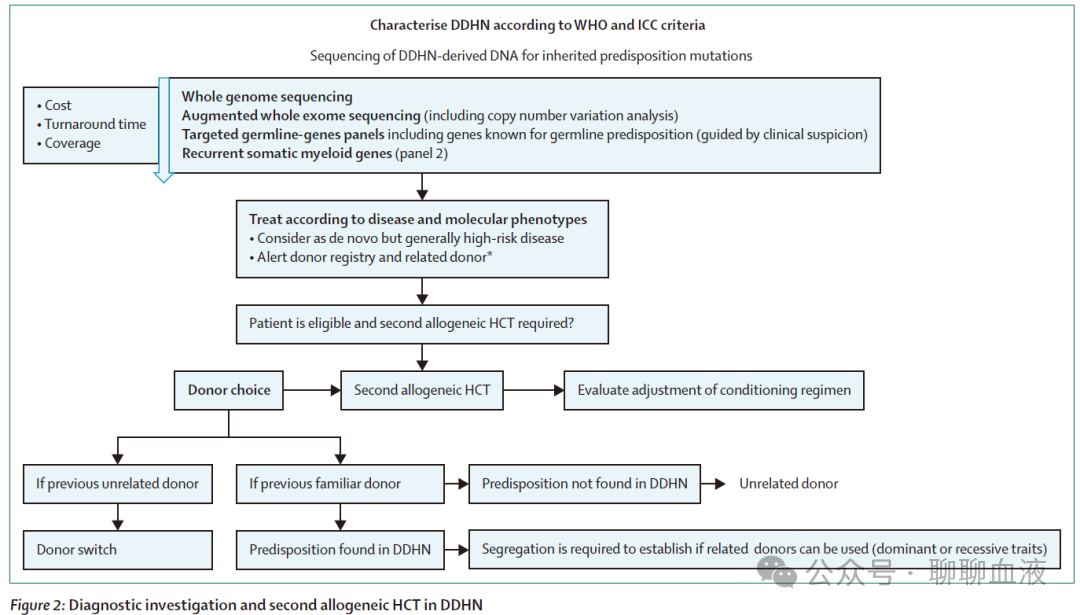
如前所述,应对所有 DDHN 进行分子嵌合体状态检测,这是确立诊断的基石。额外的检查取决于疾病特征,并应遵循 WHO 和 ICC 诊断分类。所有移植后有复发证据的髓系疾病患者应使用完整的 NGS panel(即,不限于检测移植前存在的突变)和流式细胞术(如适用)进行仔细评估。在髓系以及淋系 DDHN 中,胚系增强的体细胞 NGS panel可检测到目前已知的大部分可导致 DDHN 的易感致病基因变异(包括髓系疾病中最常见的体细胞突变和可演变为髓系疾病的遗传病中发现的胚系突变)(panel 2),以及在髓系疾病中常见的获得性体细胞突变。对于在靶向 panel 中未发现易感变异的病例,可进行全基因或外显子组测序分析(图 2)。
panel 2:DDHN 诊断时涉及的基因列表
最常见的复发性胚系易感基因
ATG2B/SKIP, ANKRD26, CEBPA, CHEK2, DDX41, ERCC6L2, ETV6, Fanconi 贫血基因 (FANCA, FANCB, FANC, FANCD2 等), GATA2, RUNX1, SAMD9, SAMD9L, SBDS, 端粒生物学基因 (DKC1, PARN, RTEL1, TERC, TERT, TINF2 等), TP53, XPD, 和 XRCC3.
复发性体细胞髓系基因
表观遗传修饰:DNMT3A, TET2, IDH1/2, WT1, EZH2, ASXL1, 和 KMD6A
RNA 剪接:SF3B1, SRSF2, 和 ZRR2
DNA 复制:STAG2, RAD21, ATM, NMP1, SETBP1, 和 DDX41
转录因子:ETV6, GATA2, CEBPA, BCOR, BORL1, 和 CUX1
细胞信号传导和生长:FLT3, KIT, JAK2, MPL, PTPN11, NRAS, KRAS, CBL, GNAS, PTEN, 和 TP53
供者管理
尽管考虑到最近获得的遗传基因组学,迫切需要适当解决干细胞捐献的伦理影响,但目前尚无超国际化指南告知如何处理此类病例。因此专家组将此问题确定为科学学会进一步讨论的主题,并提供以下建议。
所有供者应在捐献前被告知:(1)如果在捐献后发生血液病,需要提醒医疗团队;(2)可能需要对他们的捐献细胞(在冻存样本或受者体内)进行基因组分析。应征得他们同意,了解他们是否希望被告知受者体内可能对其自身产生潜在临床后果的临床或遗传发现。所有潜在供者应被询问家族性血液病史,并在捐献前评估血细胞计数。在受者有血液病遗传倾向的情况下,应为其家庭组织特定的遗传咨询。
在诊断为 DDHN 的情况下,应通过国际非亲缘供者登记处的框架通知负责该供者的团队。应根据国家法律联系供者,并在捐献前获取知情同意,送往遗传咨询并充分考虑伦理问题。建议对发生 DDHN 的患者的供者进行血细胞计数检测,以排除明显的血液病。
DDHN 的治疗
一般来说,由于预后不良,建议对所有 AML 和大多数MDS病例进行 DDHN 治疗,即使在血细胞减少轻微且患者状况允许治疗的情况下也是如此。鉴于 DDHN 的异质性和缺乏强有力的循证指南,管理应根据患者和疾病特征进行个体化。治疗决策通常应遵循标准方法,评估可操作的分子靶点(例如,NPM1, FLT3 和 IDH1/2),并在适当时纳入靶向治疗。
治疗选择必须考虑既往治疗,特别是既往蒽环类药物暴露史和距离异基因 HCT 的时间间隔,因其会影响对强化方案的耐受性。移植后早期病例或有大量合并症的患者通常需要强度较低的策略。不良遗传特征很常见,可能导致治疗耐药。
对于在首次异基因 HCT 后足够长的时间后发生的、fit的供者细胞来源 AML 患者,推荐强化化疗。对于其他 AML 患者,可考虑使用去甲基化药物联合或不联合维奈克拉。当存在可靶向突变时,应使用靶向治疗。如果患者身体状况允许,淋系恶性肿瘤应根据标准的de-novo肿瘤建议进行管理。
第二次异基因 HCT 的指征
第二次异基因 HCT 是 DDHN 的潜在治愈性选择,应在可行时提供。在大多数情况下,应在达到缓解的患者中进行,但对于惰性疾病(例如成熟淋巴细胞恶性肿瘤,以及某些骨髓增生异常综合征或慢性粒-单核细胞白血病),可考虑直接进行移植。距离首次异基因 HCT 时间短可能限制资格。在进行下一步前,必须仔细评估器官功能、合并症和整体健康状况。
尽管 DDHN 预后较差,但应考虑对所有供者来源的髓系肿瘤进行移植,并与患者明确讨论风险和获益。
第二次异基因 HCT 的供者选择
建议为第二次异基因 HCT 选择不同的供者。此外,当怀疑或确认存在胚系易感性时,使用亲缘供者必须遵循特定考虑因素(图 2)。如果使用过亲缘供者,即使胚系易感性筛查测试为阴性,仍应鼓励使用非亲缘供者,因为检测可能会遗漏潜在的未识别或未经测试的特征。对于已确定特定遗传性疾病的病例,分离研究将根据相关胚系疾病的遗传模式指导是否可以使用其他家庭成员作为供者。如果首次异基因 HCT 使用了非亲缘供者,建议选择不同的非亲缘供者。
即使在老年供者中克隆性造血更常见,也不建议仅因此原因在选择算法中排除供者,因为克隆性造血转移后 DDHN 的发生率仍然非常低,且不应常规在供者中搜索克隆性造血。然而若有多个供者可用,建议优先选择较年轻的供者。
预处理方案
建议对大多数患者使用减低毒性预处理方案。例外情况可能包括非常年轻、fit、距离首次异基因 HCT 时间较长且没有治疗相关毒性遗传易感性的个体。
减低强度预处理可减轻毒性并降低移植相关死亡率(在登记处分析中仍然非常高)。预处理策略进一步影响白血病发生,必须根据既往治疗暴露、首次异基因 HCT 间隔时间、患者健康状况、年龄和合并症进行调整。清髓性方案和放疗可诱导 DNA 损伤和基因组不稳定,应仔细评估其使用,特别是在有胚系易感综合征的患者中。
移植后管理
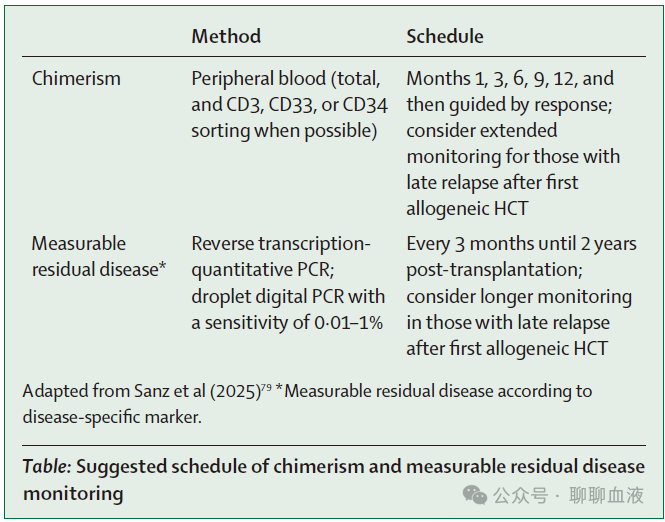
移植后维持和免疫调节策略也应予以考虑,并根据个体情况进行调整,以平衡复发和移植物抗宿主病的竞争风险。正如预期,目前仍缺乏指导具体管理的文献,在实践中,应使用标准的中心经验性程序,通常从首次异基因 HCT 推断而来。建议进行定期疾病评估,如有可能,应使用 EBMT 实践协调与指南委员会先前报道的可测量残留病监测,并进行系列嵌合体检测,包括分选细胞(CD3+、CD33+ 或 CD34+)检测(如有可能),尤其是在第二次异基因 HCT 后的前两年内(表)。
对于伴有FLT3 突变的 AML,建议采用 FLT3 抑制剂的维持治疗。对于 BCR::ABL 阳性急性淋巴细胞白血病或慢性髓系白血病,推荐移植后使用酪氨酸激酶抑制剂。由于缺乏数据,不建议在没有可用靶向治疗的患者中进行维持治疗,也不建议进行预防性供者淋巴细胞输注;但可根据标准实践,根据嵌合体和可测量残留病结果指导供者淋巴细胞输注。
接受第二次异基因 HCT 的患者发生器官毒性的风险更高,应至少进行心脏、肾脏和肝脏监测。长期存活者发生继发性恶性肿瘤的风险也更高,可能受益于系统监测和远期效应筛查,特别是当遗传性疾病带来血液外疾病风险时。
未来展望
EBMT 登记处和其他国际移植登记处对于定期、大规模前瞻性跟踪 DDHN 的发病率至关重要,同时也能记录完整的 DDHN 分子注释(包括胚系易感性)。在国际层面,建立一个统一的 DDHN 报告数据平台,并提供标准化的病例管理指南,可能会有所帮助。为了识别移植时未知的潜在遗传易感性,应推动储存供者和受者细胞。此外,应努力更好地告知供者和受者关于通过血液或骨髓传播的可能遗传性疾病,包括在捐献或移植前可能仍未知的易感条件。最后,需要进行获取供者和受者样本的转化研究,以更好地阐明 DDHN 的发病机制,特别是在遗传性和体细胞突变之间的相互作用方面。
参考文献
Lancet Haematol . 2026 Jun;13(6):e408-e417. doi: 10.1016/S2352-3026(26)00046-3.
小提示:本篇资讯仅在梅斯医学APP中开放阅读,请扫描二维码直接下载APP
