首页 > 疾病防控/ 正文
深度解析医学证据,lxfs.net为你支撑决策
论坛导读:大脑皮层发育畸形( MCD)是一组由于遗传、感染、血管或代谢等多种病因导致的大脑皮层形成障碍的罕见疾病。这类疾病虽然发病率不高,但临床负担极为沉重:患者常表现为发育迟缓、脑瘫或癫痫,其中约40%~50%的儿童药物难治性癫痫手术病例由MCD引起。由于MCD种类繁多、影像表现复杂、遗传机制多样,临床实践中长期存在术语使用不精确、分类标准不统一的问题。不同亚型之间的边界模糊,同一病变在不同文献中可能被赋予不同名称,严重阻碍了影像科医师、临床医师、病理学家和遗传学家之间的有效沟通。
大脑皮质发育畸形(Malformations of Cortical Development, MCD)是一组罕见的结构性脑疾病,但其临床影响却远非“罕见”二字所能概括。据估计,儿童难治性癫痫中约40%~50%需接受手术治疗,而MCD正是其中最主要的病因。这些患者常表现为发育迟缓、脑性瘫痪或癫痫,其神经功能结局因畸形类型、范围及所涉及的基因通路而异。
MCD的诊断长期面临一个核心困境:病理组织是诊断的金标准,但除了接受癫痫手术或尸检的患者外,病理标本几乎不可获得。这意味着,在绝大多数情况下,临床决策必须依赖于神经影像学——而影像学的准确解读,又高度依赖于统一、规范的术语体系和分类框架。正是在这一背景下,欧洲脑畸形网络Neuro-MIG(COST Action CA16118)组织了一个由国际小儿神经放射学专家组成的委员会,联合遗传学家、小儿神经科医生和病理学家,经过系统性文献回顾和多学科共识讨论,于2020年在Brain杂志上发表了题为《Definitions and classification of malformations of cortical development: practical guidelines》的共识性指南。

MCD的遗传学进展与分子机制
迄今已报道逾100个基因与MCD相关,涵盖细胞周期调控、细胞骨架功能、细胞迁移和基底膜完整性等多个生物学通路。其中几个重要通路值得特别关注:
-
mTOR信号通路是FCD II型、结节性硬化症和巨脑畸形谱系的共同分子基础。PIK3CA、AKT3、MTOR等基因的体细胞嵌合激活突变导致细胞异常增殖和分化,其表型取决于突变发生的时间和累及的细胞范围——早期广泛的突变导致HMEG,晚期局限的突变导致FCD IIb。
-
微管蛋白及相关基因(TUBA1A、TUBB2B、TUBB3、DYNC1p等)构成“微管蛋白病”谱系,可表现为无脑回、PMG样畸形或发育不良,常伴胼胝体发育不全和脑干/小脑发育不全。其病理机制涉及神经元迁移和轴突导向的双重障碍,反映了微管在细胞骨架动态中的核心作用。
-
Reelin信号通路(RELN、VLDLR)缺陷导致无脑回伴小脑发育不全,表现为前部重于后部的皮层增厚伴重度海马和小脑发育不全。
-
FLNA突变是X连锁弥漫性室周结节性异位的最常见原因,女性杂合子呈典型表现(异位结节沿侧脑室外侧壁分布),男性半合子通常胚胎致死或表型更重。
-
DCX(双皮质素)突变导致X连锁无脑回/带状异位,男女表型差异显著——女性杂合子表现为SBH,男性半合子表现为前部显著的无脑回。
-
体细胞嵌合突变在多种MCD中具有重要地位,包括HMEG、FCD II型和部分无脑回畸形。这一发现改变了MCD“全脑性遗传病”的传统观念,提示了针对突变细胞群体进行靶向干预的潜在可能性。
-
系统综述揭示了一个关键知识缺口:在132个与MCD相关的基因中,仅有46个(34.8%)有神经病理学数据报道,且多数来自胎儿终止妊娠后的尸检材料。该研究建立的四分类系统(具明确基因型-病理学关联的基因13个、证据有限的基因27个、存在冲突证据的基因5个、无病理数据的基因87个)清晰表明了当前知识的不均衡性,强调标准化病理评估对理解MCD发病机制的重要价值。
从胚胎发育理解MCD:分类的逻辑起点
理解MCD的分类,首先需要理解大脑皮质是如何形成的。人脑皮质的发育可大致分为三个阶段:神经增殖、神经元迁移和迁移后发育。
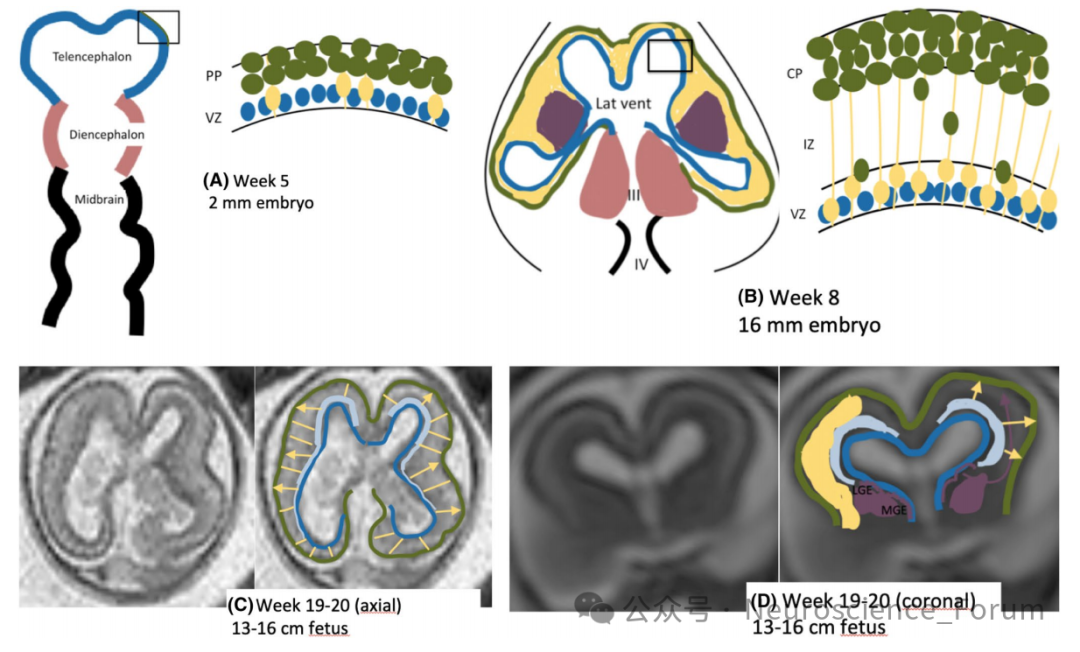
神经增殖阶段发生在生发基质中,位于侧脑室周围的室管膜下区。顶端放射状胶质细胞(apical radial glial cells, aRGC)作为神经干细胞,通过对称分裂扩增祖细胞池,再通过不对称分裂产生神经元。这一过程的异常可导致小头畸形(神经元过少)或巨脑畸形(神经元过多)。
神经元迁移是MCD分类中最核心的环节。新生神经元需从室管膜下区迁移至大脑皮质的最终位置。迁移方式主要有两种:放射状迁移——神经元沿放射状胶质细胞纤维从深部向表层移动;切向迁移——主要用于中间神经元的迁移。迁移过程的异常可表现为:神经元完全无法迁移(形成室周结节性异位)、迁移中途停滞(形成皮质下带状灰质异位)、迁移过度进入软脑膜(形成鹅卵石样畸形)等。
迁移后发育阶段则涉及轴突发生、树突生长、突触形成与修剪,以及皮质折叠(脑回与脑沟的形成)。这一阶段的异常可导致多小脑回畸形、局灶性皮质发育不良等。
这三个阶段并非截然分开,而是遗传和功能上相互依存的。同一个基因可能在不同阶段发挥作用,不同基因也可能通过同一通路导致相似的影像学表型。这为MCD的分类带来了根本性的挑战——也是这篇指南试图回应的核心问题。
关键MCD亚型的影像学定义
2020年指南对MCD各亚型给出了清晰的影像学定义和诊断要点,以下逐一梳理。
(一)小头畸形与巨脑畸形
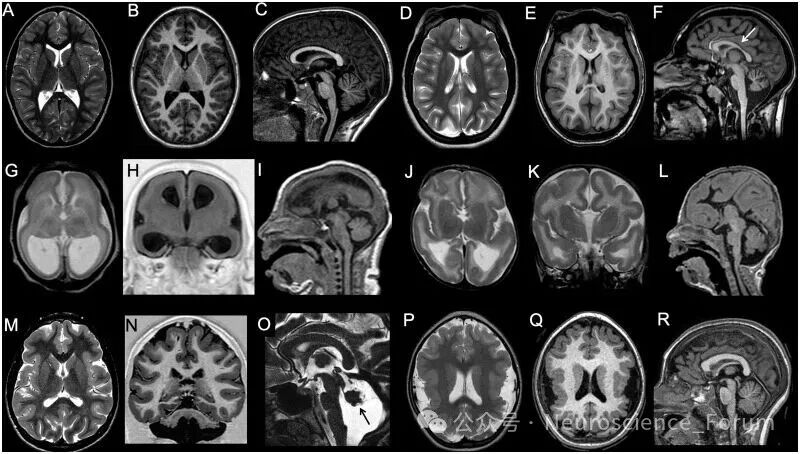
小头畸形指枕额围低于同年龄同性别均值2个标准差以上。影像学上需进一步区分:皮质正常者、简化脑回模式者(脑沟浅、脑回减少但皮质厚度正常)、伴无脑回畸形者(皮质增厚伴脑回缺失)、伴多小脑回或灰质异位者。小头畸形伴无脑回畸形(曾称“微小无脑回畸形”)并非单一疾病,而是一组异质性 disorders,常伴严重的小脑发育不全。
巨脑畸形指大脑体积增大。值得注意的是,巨头畸形不等于巨脑畸形——前者还可能由脑积水、颅外间隙增宽或骨骼发育不良引起。巨脑畸形可分为双侧对称性、双侧不对称性和单侧性(即半侧巨脑畸形)。半侧巨脑畸形若仅累及部分脑叶(如额叶或顶枕叶),则被称为“叶状半侧巨脑畸形”或“象限性发育不良”。近年来研究发现,巨脑畸形与局灶性皮质发育不良II型同属mTOR通路异常导致的谱系疾病。
(二)局灶性皮质发育不良
局灶性皮质发育不良(Focal Cortical Dysplasia, FCD)是MCD中最具临床重要性的亚型之一,也是药物难治性癫痫的主要手术适应证。
2011年国际抗癫痫联盟将其分为三型:
I型:皮质柱状(Ia)或层状(Ib)结构异常,或两者兼有(Ic)。影像上通常表现为微妙的皮质-白质分界模糊,常难以发现。
II型:皮质分层明显紊乱,伴形态异常的细胞——IIa型仅含异形神经元,IIb型同时含异形神经元和气球样细胞。影像上典型表现为局灶性皮质增厚或变薄、脑回模式异常、皮质下白质FLAIR/T2高信号,以及最具特征性的穿脑征(transmantle sign)——自皮质向侧脑室方向呈漏斗状延伸的白质高信号。
III型:I型FCD合并同侧脑叶的其他病变(海马硬化、肿瘤、血管畸形或早期获得性损伤)。
指南特别指出,结节性硬化症的皮质结节应被视为FCD IIb的一个亚型,因两者在组织学、影像学和遗传学(均涉及mTOR通路)上高度相似。此外,新生儿期FCD的MRI信号特征与年长儿相反——在T2上呈低信号、T1上呈高信号,可能与局部髓鞘化加速有关。
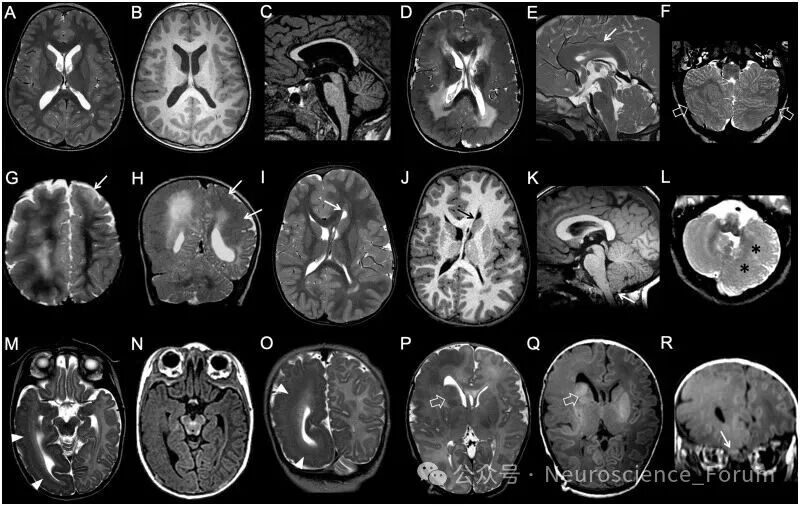
(三)灰质异位
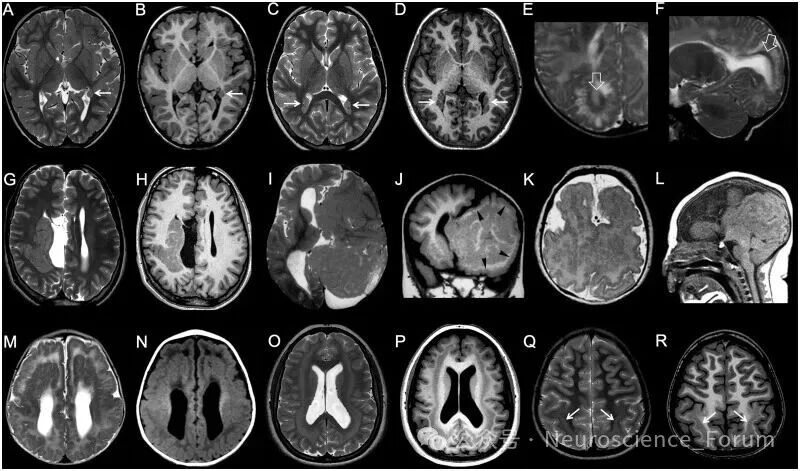
灰质异位指正常神经元在异常位置的聚集,主要源于神经元迁移障碍。根据形态和位置可分为:
室周结节性异位:最常见类型,表现为沿侧脑室壁排列的灰质信号结节。需与结节性硬化症的室管膜下结节鉴别——后者与灰质非等信号、常钙化、可强化,且垂直于室壁。
皮质下异位:白质内的灰质团块,可呈柱状穿通全层(穿通性异位)或大块卷曲状。
皮质下带状异位(又称“双皮质”):位于皮质下白质内的平滑带状灰质,与上覆皮质和下位脑室之间均有正常白质分隔。可分为厚带(>7mm,位于深部白质)和薄带(1-7mm,位于浅部白质)。
(四)无脑回畸形与鹅卵石样畸形
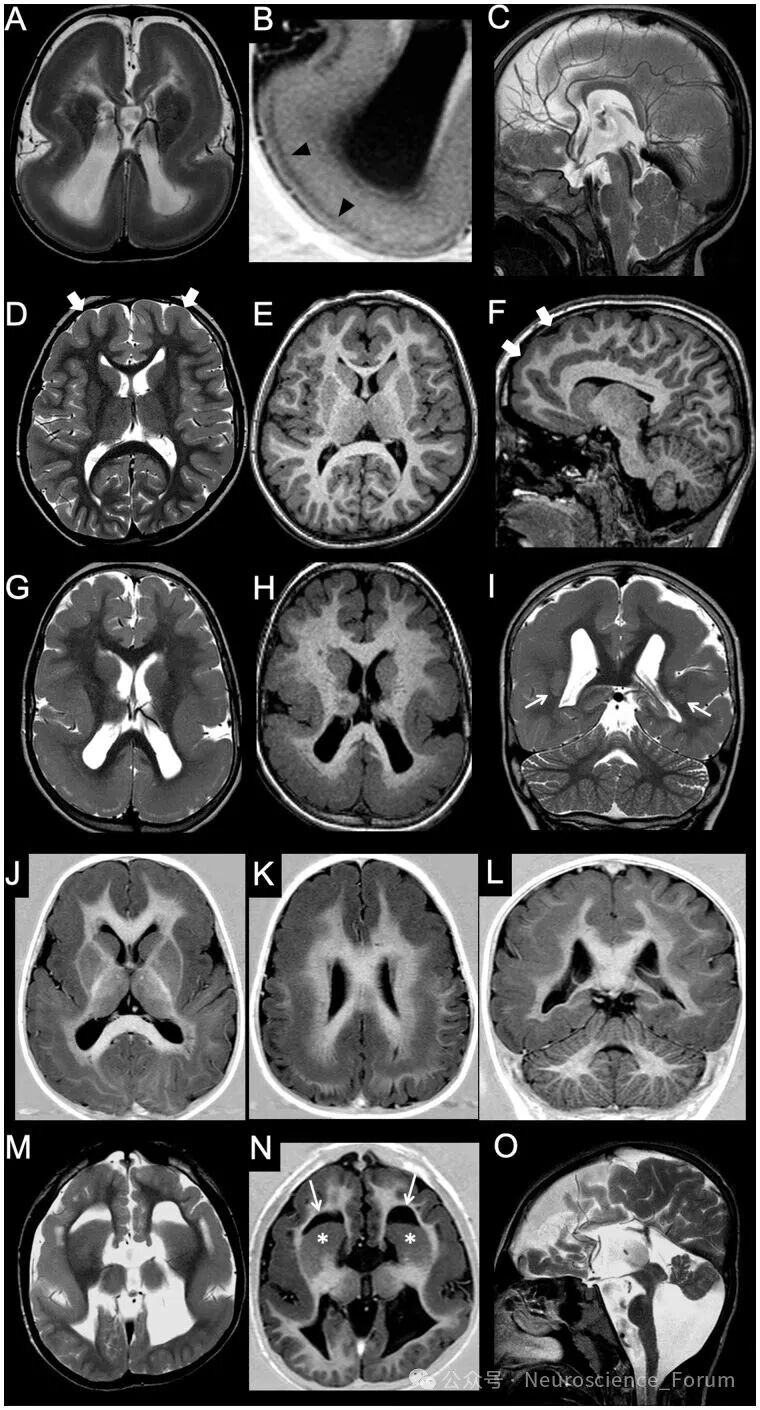
无脑回畸形字面意为“光滑脑”,核心影像学特征为皮质增厚伴脑回减少。严重者呈“8字征”——完全无脑沟、外侧裂宽大垂直。根据皮质厚度可分为厚型(10-20mm,常见四层结构)和薄型(5-10mm)。无脑回畸形常伴前或后部优势的梯度分布,这有助于预测致病基因。
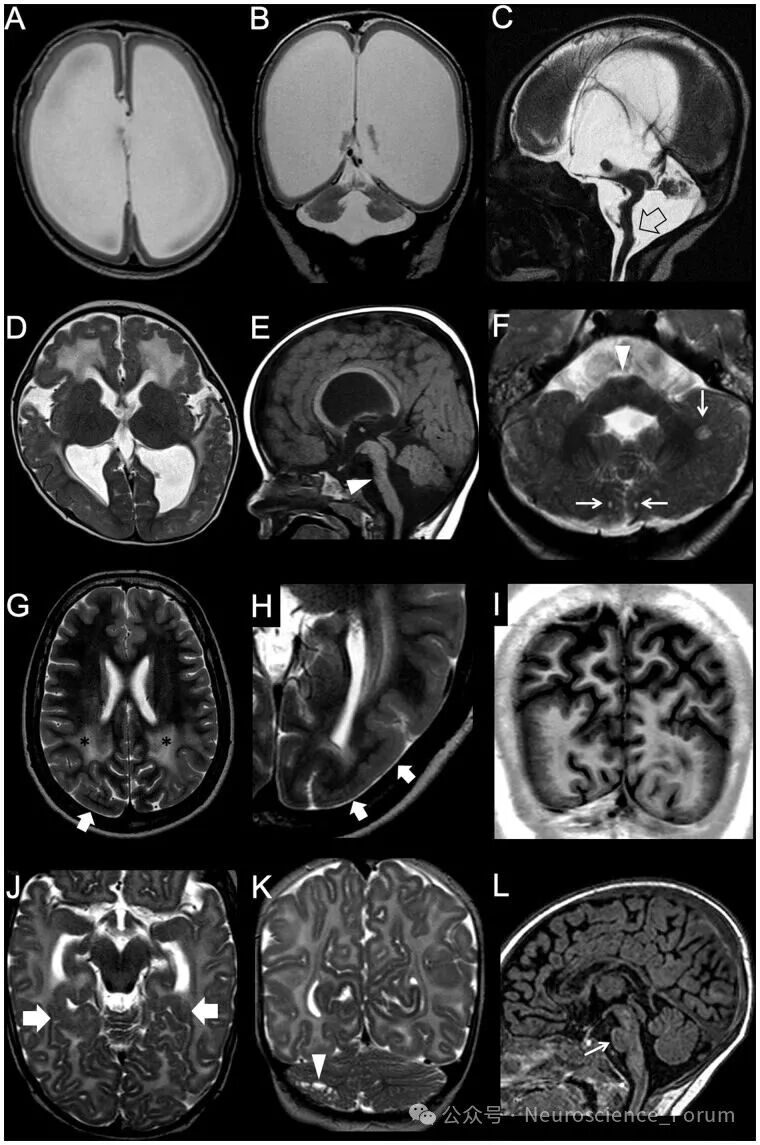
鹅卵石样畸形(曾称II型无脑回畸形)的病理机制与无脑回畸形截然不同——后者是神经元迁移不足,而前者是神经元过度迁移进入软脑膜下腔。影像上表现为皮质表面不规则的“鹅卵石”样外观,皮质-白质边界呈锯齿状,常伴垂直条纹和下方白质信号异常。
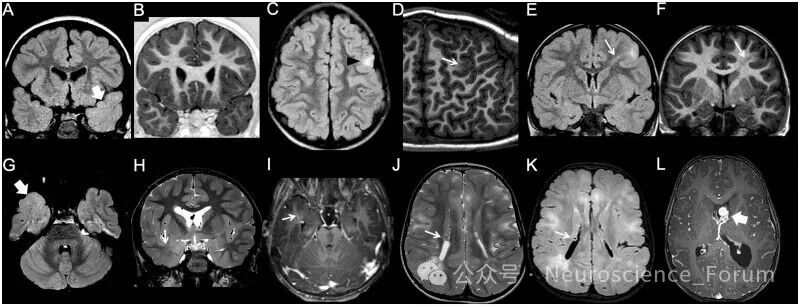
(五)多小脑回畸形与裂脑畸形
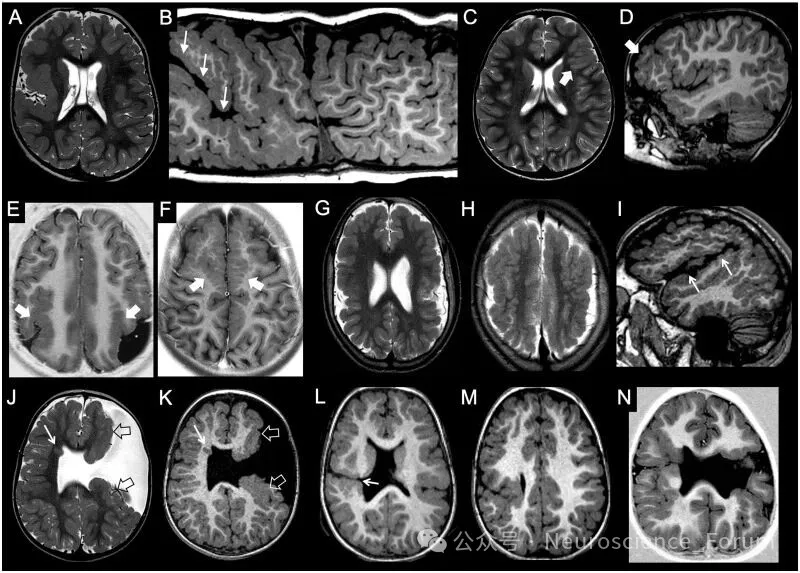
多小脑回畸形指异常增多的小脑回,可局灶、多灶或弥漫,单侧或双侧。最常见累及外侧裂周围(约60%-70%)。影像表现因髓鞘化阶段而异:未髓鞘化脑呈薄而起伏的“点彩状”灰白质交界,髓鞘化后则表现为增厚的“光滑”皮质。多小脑回畸形与鹅卵石样畸形构成影像谱系——取决于软脑膜限界膜缺损的大小。
裂脑畸形指贯穿大脑半球全层的裂隙,衬以多小脑回样灰质。分为开唇型(裂隙开放、充满脑脊液)和闭唇型(裂隙闭合,脑室侧可见“凹陷”)。
(六)脑回结构不良
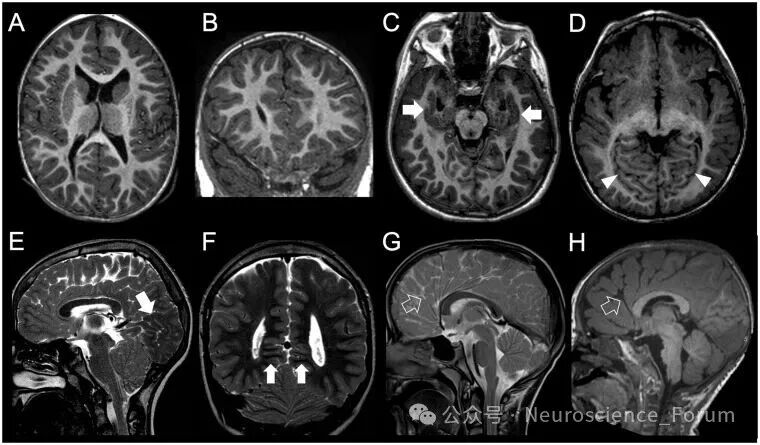
脑回结构不良是近年来提出的新术语,指皮质厚度正常但脑回模式异常——脑沟深度和方向异常,但不符合多小脑回、无脑回或单纯脑回简化的典型特征。最初在微管蛋白基因突变和肌营养不良蛋白聚糖病中被描述。影像上需注意:正常人的主要脑沟和次要脑沟虽有轻微变异,但位置和深度基本恒定且对称、间距<1.5cm——超出此范围即可考虑脑回结构不良。
影像检查策略:如何“看见”MCD
2020年指南对MCD的影像检查给出了具体而实用的建议,核心要点如下:
首选MRI,因其无电离辐射且能清晰区分灰白质。推荐3T优于1.5T。髓鞘化进程对影像判读至关重要:新生儿期皮质-白质对比最佳;8-12个月的“等信号期”灰白质对比最差,MCD最易漏诊;因此,若首次MRI在1岁内完成且结果阴性但临床高度怀疑,应在3岁左右(髓鞘化基本完成)复查MRI。
序列选择需因龄而异:髓鞘化完全者应获得薄层(≤1mm)3D T1加权、轴位和冠状位T2加权(≤3mm)以及FLAIR(最好3D薄层);新生儿则以三平面T2加权最为重要,FLAIR不必要。弥散张量成像在科研和临床中均有重要价值。
辅助检查:FDG PET可帮助定位MRI阴性的致痫灶(呈低代谢);发作期SPECT或ASL灌注MRI可显示癫痫发作区的高灌注。
分类的挑战与未来方向

MCD的分类体系是“动态的、流动的模型”,而非“刚性的、不可更改的范式”。这一认识源于以下几个根本性挑战:
第一,基因型-表型的复杂性。同一基因的不同突变可导致不同类型的MCD,而同一通路中不同基因的突变又可能导致相同的影像学表型。例如,LIS1和DCX突变均可导致无脑回畸形,但梯度分布不同。
第二,分期界限的模糊化。增殖、迁移和迁移后组织在遗传和功能上相互依存。例如,多小脑回畸形传统上被归为迁移后障碍,但近年病理研究显示其常伴软脑膜限界膜缺陷和软脑膜异位,提示其本质上是晚期迁移障碍。指南明确指出,未来分类将越来越依赖于对生物学通路的精确认识。
第三,FCD分类的持续演变。指南引用了Najm等2018年对ILAE 2011年FCD分类的批评性更新。事实上,2022年ILAE工作组已正式发布了FCD分类的更新版,新增了“轻度MCD伴少突胶质细胞增生(MOGHE)”等新类别。
第四,影像与病理的差距。FCD I型在影像上常呈“阴性”;部分MCD在髓鞘化过程中会“消失”又“重现”;多小脑回畸形在不同髓鞘化阶段呈现截然不同的影像表现。这些都提醒我们:影像上的“正常”不等于大脑的正常。
临床管理与预后
大脑皮层发育畸形的临床表现可从两个维度理解。弥漫性/严重型(如重度小头畸形、无脑回畸形、双侧广泛多小脑回畸形)常于婴儿期以喂养困难、发育迟缓或癫痫起病,预后较差。局灶性/轻型(如局灶性皮层发育不良、孤立性异位)常以局灶性癫痫为首发,认知可基本正常。
癫痫是核心问题。无脑回畸形患儿35%-85%出现婴儿痉挛,局灶性皮层发育不良是儿童难治性局灶性癫痫最常见病因之一。室管膜下结节性异位本身并非固有致痫灶,但可参与广泛致痫网络,手术需立体定向脑电图引导。癫痫治疗应个体化。局灶性皮层发育不良II型手术切除皮质病灶效果良好(50%-70%无发作),无需切除经脑白质高信号区(含电静默的球样细胞)。室管膜下结节性异位可考虑立体定向脑电图引导射频热凝或激光间质热疗。药物治疗需注意局灶性皮层发育不良II型中GABA受体异常可能影响苯二氮䓬类疗效。多学科管理至关重要,需神经科、影像科、遗传科、神经外科及康复科协作。产前诊断者应于三级医疗中心分娩,生后完善高分辨MRI及遗传检测。
影响预后的关键因素包括:畸形范围(弥漫性 vs 局灶性)、对称性(双侧对称最重)、头围(<-3 SD或>+3 SD预后差)、癫痫起病年龄(<3月龄预后差)及合并畸形(胼胝体、小脑、脑干受累提示预后不良)。
结语与展望
MCD作为一组临床异质性极强的神经系统发育性疾病,正经历从形态学描述向分子机制驱动的精准分类的范式转变。整合胚胎发育生物学、高分辨率神经影像学、下一代测序技术和标准化神经病理学评估的多学科方法,不仅提高了MCD的诊断准确性,也为理解其发病机制、优化遗传咨询和探索靶向治疗策略奠定了基础。随着胎儿医学和产前诊断技术的进步,未来MCD的管理将更加强调早期识别、个体化干预和全生命周期随访。对于临床医师而言,熟悉MCD的影像学特征谱系、掌握关键基因-表型关联、了解产前诊断的最新进展,是提供高质量医疗服务的必要前提。
参考文献
-
Russ JB, et al. Fetal malformations of cortical development: review and clinical guidance. Brain. 2025 Jun 3;148(6):1888-1903. doi: 10.1093/brain/awaf094.
-
Severino M, et al. Definitions and classification of malformations of cortical development: practical guidelines. Brain. 2020 Oct 1;143(10):2874-2894. doi: 10.1093/brain/awaa174.
-
Hoeberigs MC, et al. Malformations of cortical development: Embryology and epilepsy. Epilepsia. 2025 Oct;66(10):3642-3655. doi: 10.1111/epi.18501.
-
Brock S, et al. Neuropathology of genetically defined malformations of cortical development-A systematic literature review. Neuropathol Appl Neurobiol. 2021 Aug;47(5):585-602. doi: 10.1111/nan.12696.
-
Guerrini R, Dobyns WB. Malformations of cortical development: clinical features and genetic causes. Lancet Neurol. 2014 Jul;13(7):710-26. doi: 10.1016/S1474-4422(14)70040-7.
小提示:本篇资讯仅在梅斯医学APP中开放阅读,请扫描二维码直接下载APP
