首页 > 疾病防控/ 正文
深度解析医学证据,lxfs.net为你支撑决策
背景介绍
骨质疏松症是一种以骨量减少、骨微结构破坏和骨折风险增加为特征的年龄相关代谢性骨病,严重影响全球数亿中老年人的生活质量。正常骨稳态依赖于成骨细胞介导的骨形成与破骨细胞介导的骨吸收之间的精密平衡——这一过程被称为“成骨-破骨偶联”。然而,这种偶联也带来了治疗上的巨大挑战:许多关键调控因子同时在成骨和破骨细胞中发挥功能,靶向它们可能同时抑制骨形成和骨吸收,限制了治疗窗口。Runx2作为公认的成骨细胞分化主转录因子,其全球敲除可导致完全无骨形成。但有趣的是,Runx2缺失小鼠中破骨细胞分化也完全消失,提示Runx2可能在破骨细胞中同样发挥重要功能。这一双重角色使得直接靶向Runx2治疗骨质疏松面临“顾此失彼”的困境。
研究思路
针对上述挑战,南方科技大学医学院曹惠玲教授团队系统探究了Runx2在破骨细胞中的内在功能及其调控机制。通过单细胞转录组分析,团队意外发现Runx2在造血干细胞及单核细胞(破骨细胞前体)中的表达水平甚至高于成骨谱系细胞。利用破骨细胞前体特异性Runx2敲除小鼠(Lyz2-Cre;Runx2^(fl/fl)),他们发现Runx2缺失显著抑制了破骨细胞分化、增加了骨量,并证明Runx2直接结合并转录激活破骨细胞标志基因Ctsk(编码组织蛋白酶K)的启动子。然而,由于Runx2同时调控成骨分化,直接靶向Runx2会同时抑制骨形成。为解决这一矛盾,团队通过转录组筛选鉴定出Ip6k2作为Runx2的相互作用蛋白——Ip6k2特异性增强Runx2依赖的Ctsk转录,但仅局限于破骨细胞中。机制上,Ip6k2与Runx2物理结合,可能通过协同转录共因子Fli1或Erg增强Runx2的DNA结合能力。在破骨细胞前体中敲除Ip6k2可完全模拟Runx2缺失的表型——抑制破骨细胞生成、增加骨量,且不影响成骨细胞功能。更重要的是,在去卵巢(OVX)诱导的雌激素缺乏骨质疏松模型和衰老相关骨质疏松模型中,基因敲除或小分子抑制剂UNC7467介导的Ip6k2药理学抑制均能有效保护骨量丢失。相关内容以Targeting the Ip6k2-Runx2 axis disrupts osteoblast-osteoclast coupling to treat osteoporosis发表在Nature Communications!

图片解析
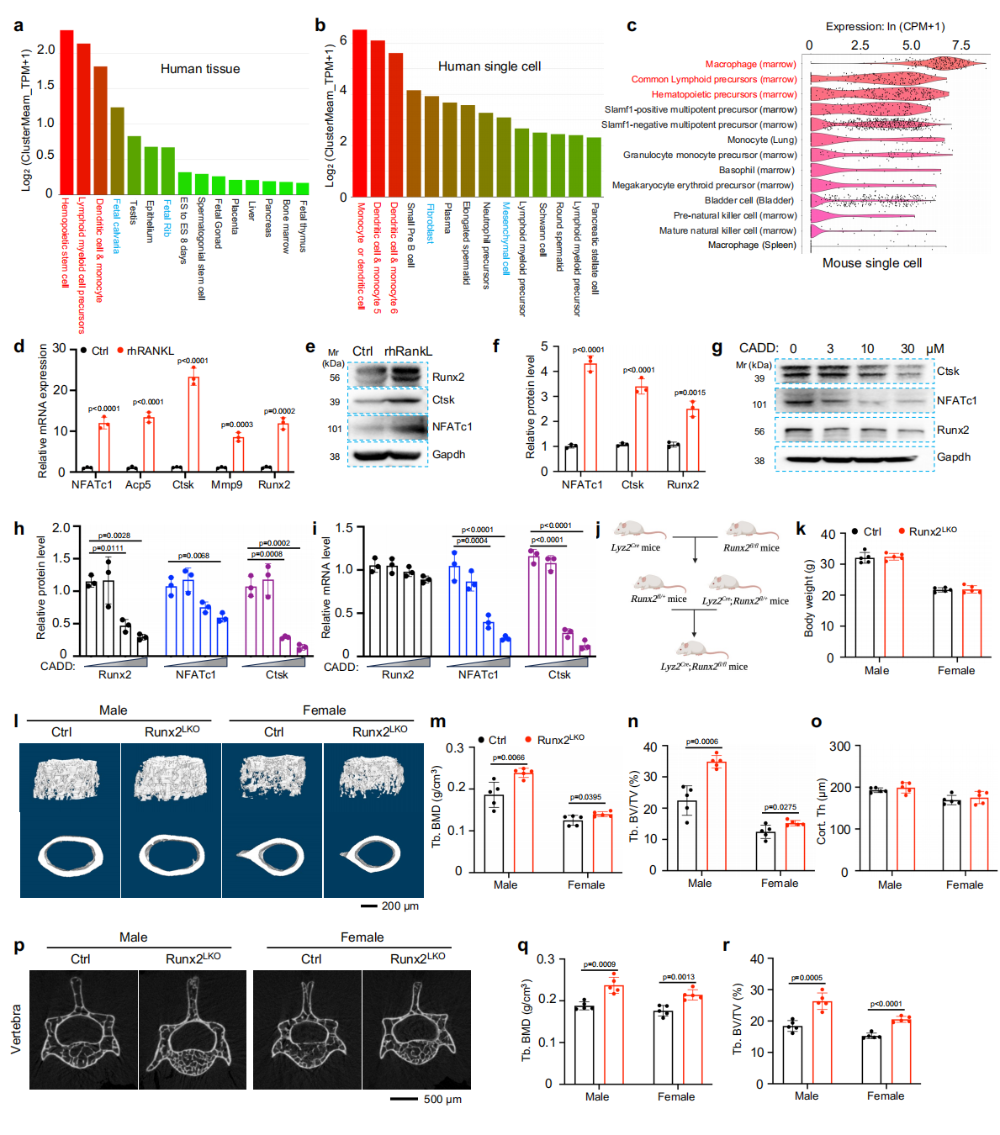
图1. Runx2在破骨细胞生成中上调,其缺失增加骨量:(a-b)人类单细胞转录组显示Runx2在单核细胞和树突状细胞中表达最高,甚至高于骨组织中的成骨谱系细胞。(c)小鼠单细胞数据同样显示Runx2在骨髓巨噬细胞中富集。(d-f)RANKL诱导破骨分化过程中Runx2及NFATc1、Ctsk等标志基因mRNA和蛋白水平显著上调。(g-i)Runx2抑制剂CADD522显著抑制破骨分化及NFATc1/Ctsk表达。(j)Lyz2-Cre介导的破骨前体Runx2敲除策略示意图。(k-r)μCT显示Runx2^(LKO)小鼠股骨远端和腰椎骨量显著增加,男女均有效。
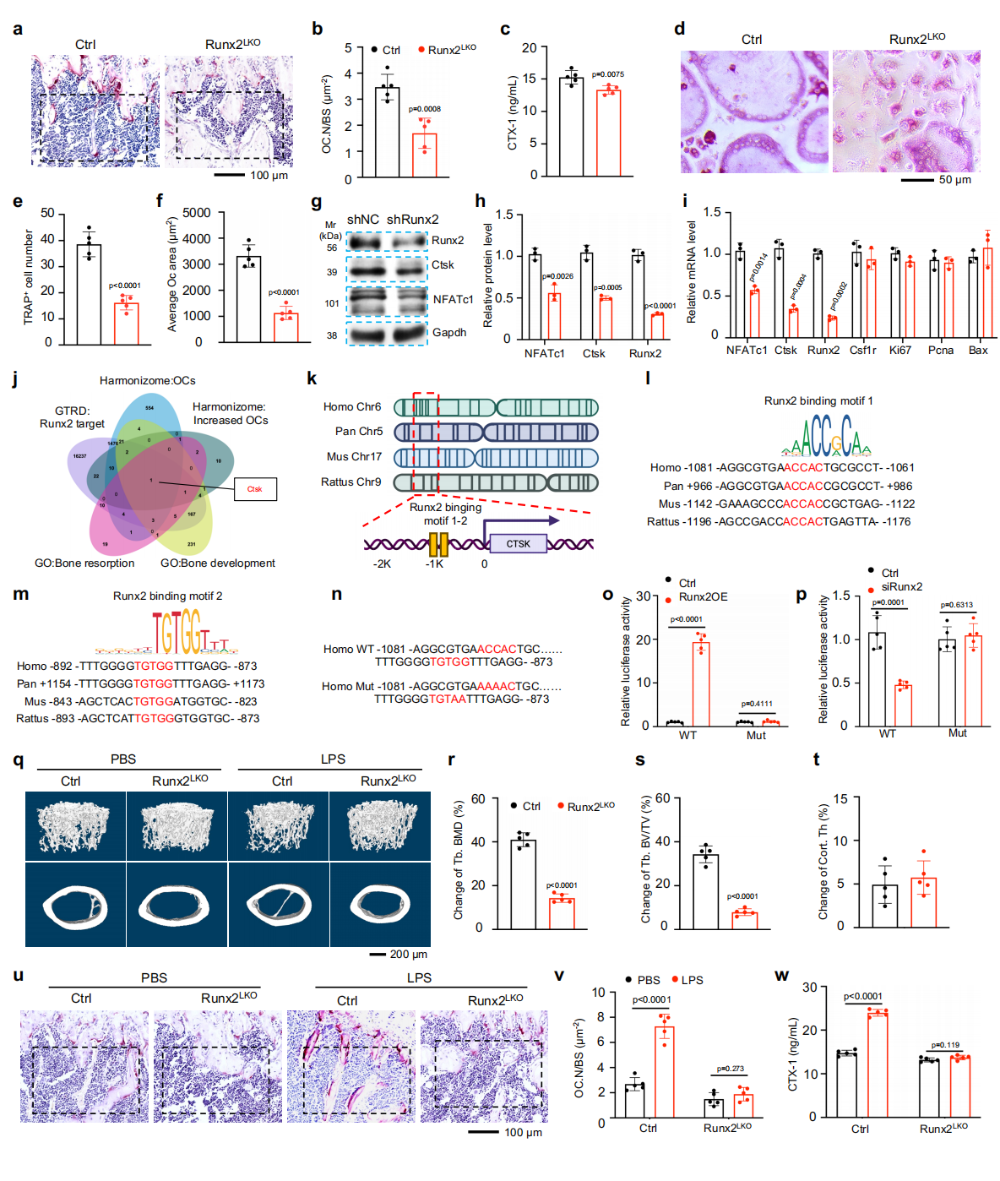
图2. Runx2直接调控破骨细胞中Ctsk转录,其缺失抵抗炎症诱导的骨丢失:(a-c)Runx2^(LKO)小鼠TRAP染色和血清CTX-1证实破骨细胞活性显著降低。(d-f)体外破骨分化实验显示Runx2^(LKO)来源BMMs分化能力受损。(g-i)shRNA敲低Runx2显著下调NFATc1和Ctsk表达,但不影响早期分化标志Csf1r、增殖标志Ki67/Pcna和凋亡标志Bax。(j)生信分析鉴定Ctsk为Runx2的直接靶基因。(k-m)跨物种比对显示Ctsk启动子含两个保守Runx2结合基序。(n-p)双荧光素酶报告基因证实Runx2直接激活Ctsk启动子,突变结合基序则消除该效应。(q-w)LPS诱导炎症模型中,Runx2^(LKO)小鼠骨丢失显著减轻,TRAP+破骨细胞和CTX-1水平均未升高。
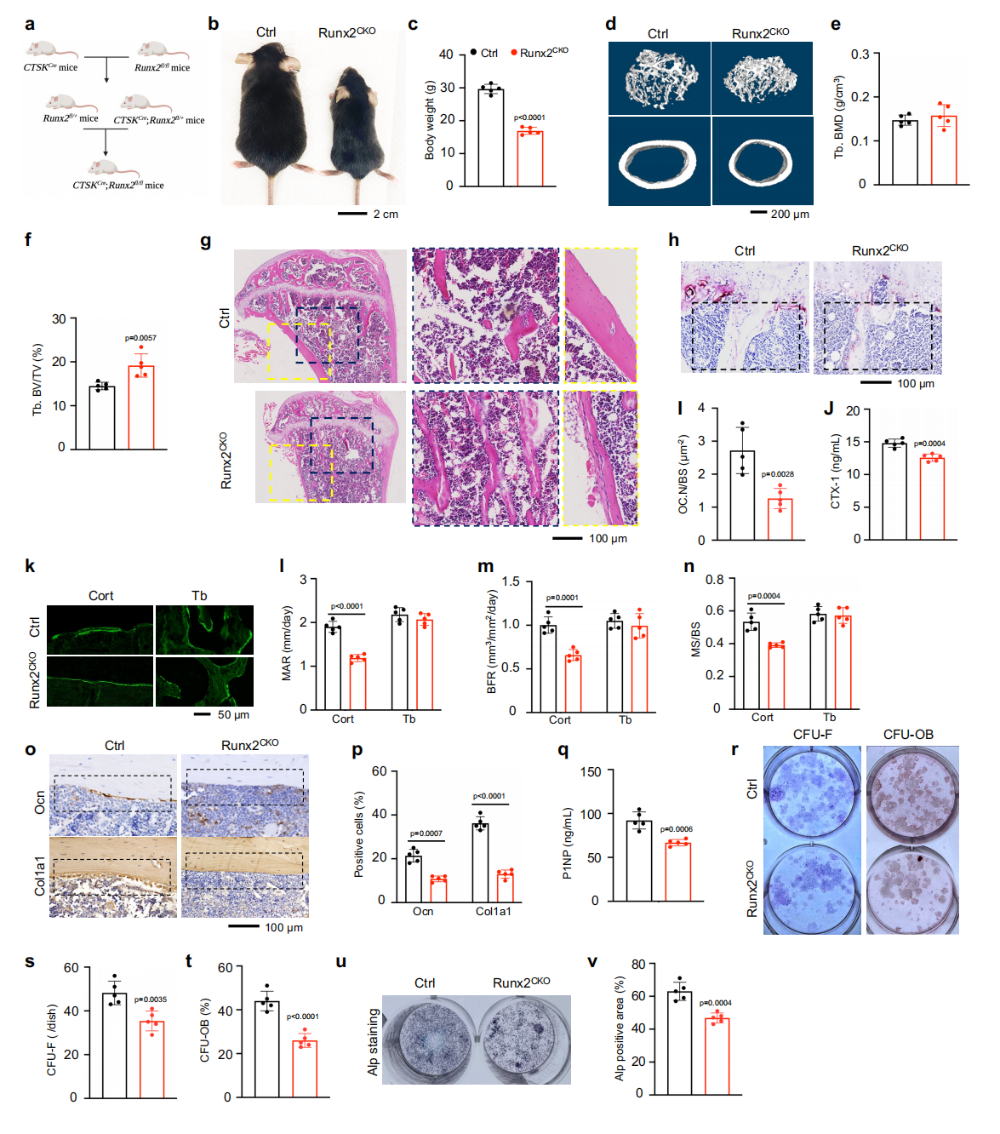
图3. Runx2通过同时调控成骨和破骨分化协调成骨-破骨偶联:(a)CTSK-Cre介导的成熟破骨细胞Runx2敲除策略。(b-g)Runx2^(CKO)小鼠体重降低、骨小梁骨量增加但皮质骨厚度显著减少。(h-j)TRAP染色和CTX-1证实成熟破骨细胞活性受损。(k-p)钙黄绿素双标和IHC显示Runx2^(CKO)小鼠皮质骨形成率显著降低,Ocn和Col1a1阳性成骨细胞减少。(q-v)CFU-F、CFU-OB和ALP染色证实Runx2缺失损害骨膜干细胞的成骨分化能力。
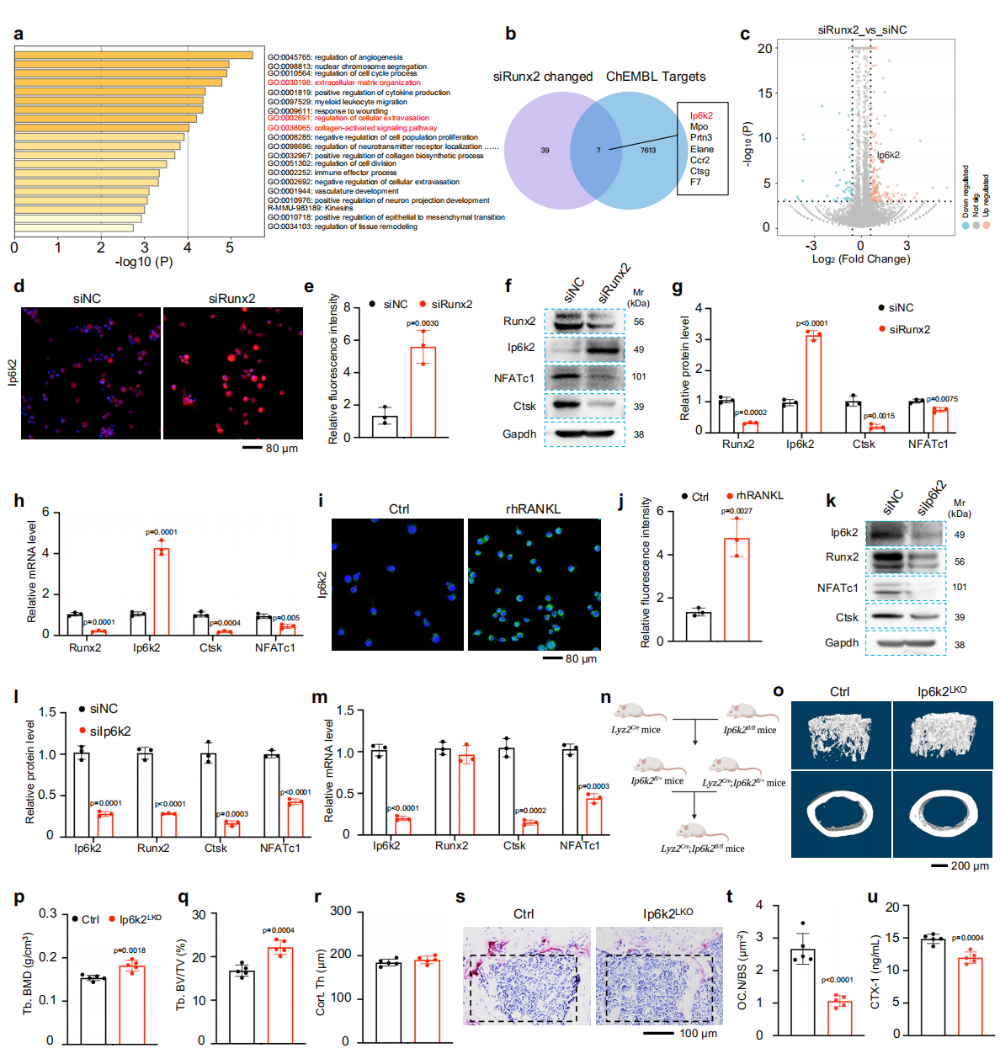
图4. Ip6k2与Runx2关联调控破骨细胞生成:(a-c)Runx2敲低后转录组分析及与ChEMBL药物靶基因交叉,鉴定出Ip6k2为候选基因。(d-h)Runx2敲低后Ip6k2蛋白和mRNA水平代偿性升高,而Ctsk降低。(i-j)RANKL刺激同样诱导Ip6k2表达上调。(k-m)Ip6k2敲低显著降低Runx2和Ctsk蛋白水平,但不影响Runx2 mRNA。(n)Ip6k2破骨前体敲除策略。(o-r)Ip6k2^(LKO)小鼠骨小梁骨量显著增加,皮质骨无变化。(s-u)TRAP染色和CTX-1证实破骨细胞活性降低。
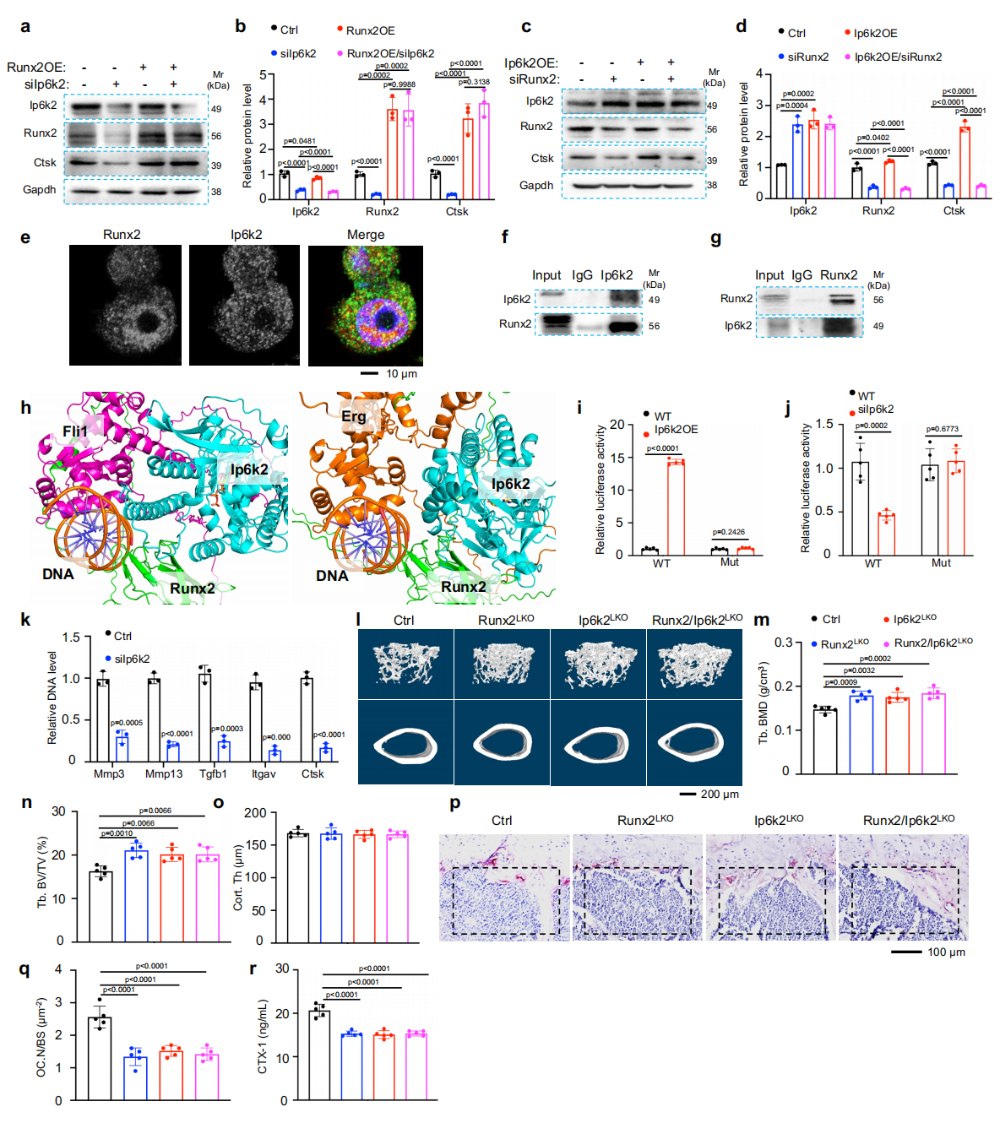
图5. Ip6k2对破骨细胞生成的调节由Runx2介导:(a-b)过表达Runx2可完全挽救Ip6k2敲低引起的Ctsk下降。(c-d)而过表达Ip6k2无法挽救Runx2敲低引起的Ctsk下降,证明Ip6k2位于Runx2上游。(e)IF显示Ip6k2与Runx2在细胞内共定位。(f-g)Co-IP证实二者存在内源性物理相互作用。(h)AlphaFold3预测Ip6k2-Runx2-Fli1/Erg复合物结构。(i-j)双荧光素酶报告基因显示Ip6k2增强Ctsk启动子活性,突变Runx2结合基序后该效应消失。(k-n)Runx2/Ip6k2双敲除小鼠骨量增加与单敲除相当,无叠加效应。(o-q)TRAP染色和CTX-1证实双敲除组破骨活性降低程度与单敲除一致。
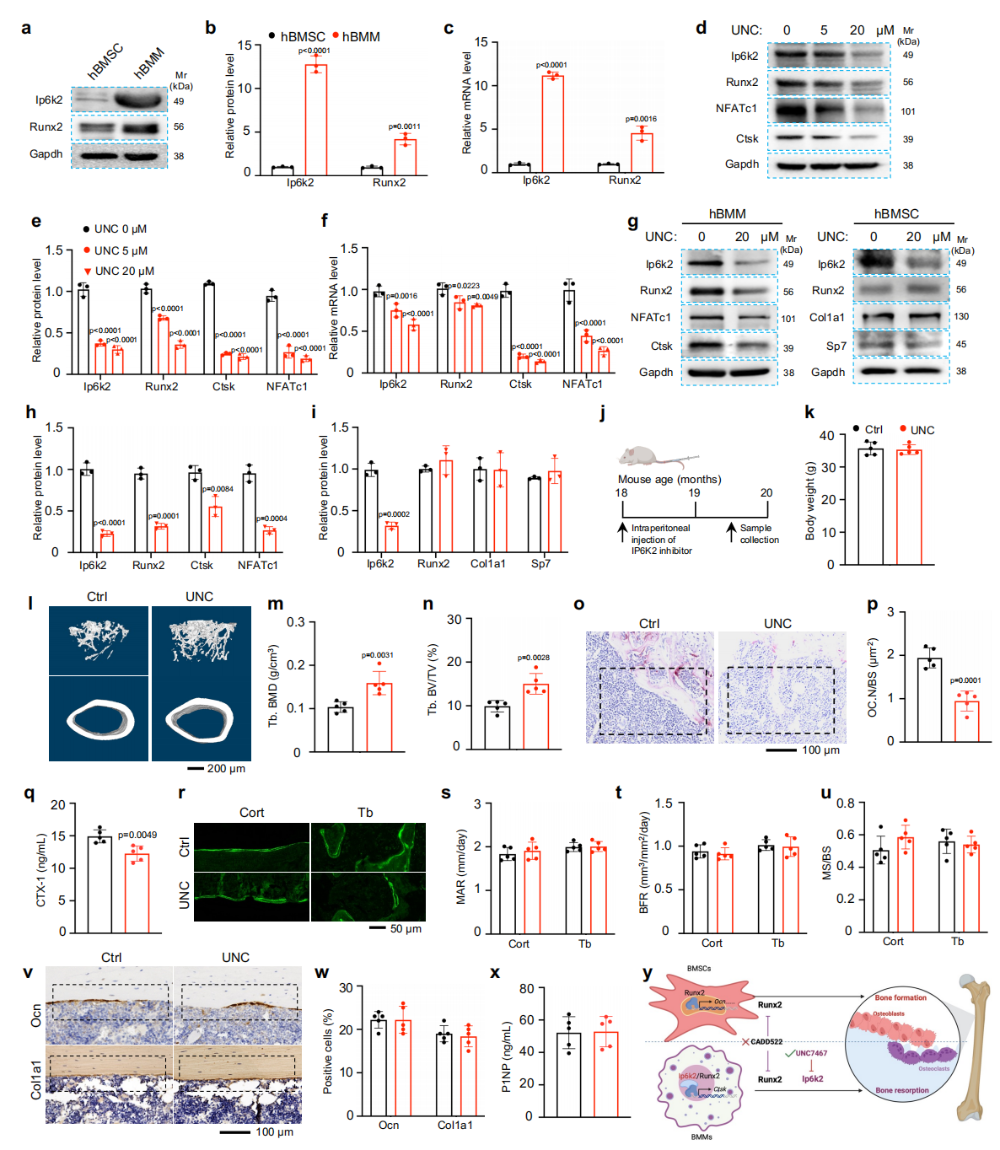
图6. Ip6k2选择性调控破骨活性,是骨质疏松治疗的潜在靶点:(a-c)人BMMs中Ip6k2和Runx2表达显著高于BMSCs。(d-f)UNC7467处理剂量依赖性降低Ip6k2、Runx2、NFATc1和Ctsk蛋白水平,但不影响Runx2 mRNA。(g-i)UNC7467抑制人BMMs破骨分化但不影响hBMSCs成骨分化。(j)衰老小鼠(18月龄)UNC7467给药方案。(k-n)UNC7467治疗6周后骨小梁骨量增加约50%,体重无变化。(o-q)TRAP染色和CTX-1证实破骨活性降低。(r-x)钙黄绿素双标、IHC和P1NP ELISA显示成骨细胞活性未受影响。(y)机制模型总结:Runx2在成骨细胞中促进骨形成,在破骨细胞中与Ip6k2协同促进Ctsk转录和骨吸收;靶向Ip6k2可选择性地抑制破骨细胞而不影响成骨细胞,实现成骨-破骨解偶联治疗。
结论
本研究系统揭示了Runx2在破骨细胞中的内在功能及其调控机制,并发现Ip6k2作为Runx2的破骨细胞特异性互作蛋白,为骨质疏松治疗提供了“解偶联”新策略。机制上,Runx2直接结合并转录激活Ctsk启动子,促进破骨细胞分化和骨吸收;而Ip6k2通过与Runx2物理结合、增强其转录活性,特异性调控破骨细胞中的Runx2功能。在破骨前体中敲除Runx2或Ip6k2均可抑制破骨生成、增加骨量,且双敲除无叠加效应,证明二者处于同一信号通路。更重要的是,Ip6k2在破骨细胞中高表达、在成骨细胞中低表达的特性,使其成为理想的药物靶点——基因敲除或小分子抑制剂UNC7467介导的Ip6k2抑制均能有效保护雌激素缺乏和衰老相关的骨量丢失,且不影响成骨细胞功能。该研究首次提出了通过靶向Ip6k2-Runx2轴实现成骨-破骨解偶联的治疗策略,为骨质疏松症的药物研发提供了全新方向。
小提示:本篇资讯仅在梅斯医学APP中开放阅读,请扫描二维码直接下载APP

猜你喜欢
- 19家医药企业被开罚单,竟查出了这些“猫腻”!
- 《中国年龄相关性黄斑变性临床诊疗指南(2023年)》发布,阿柏西普眼内注射溶液主动治疗和延长(T&E)方案获益更佳
- 防疫都了解:会议室防疫要留意这种关键点
- 科学突破!宁夏医科大学重大发现:揭秘炙甘草如何神奇治愈心律失常!
- 男性肾移植后遗精的处理方法有哪些 介绍男性肾移植后遗精的应对方法
- 雪菜鸡丝面的做法-家常味煮菜谱
- 老人经常会觉得小腿痛是什么原因
- 世卫组织:全球累计新冠确诊病例达54301156例
- Lancet:他拉唑帕尼加恩杂鲁胺治疗一线转移性去势抵抗性前列腺癌 (TALAPRO-2) 患者:一项随机、安慰剂对照的 3 期试验
- 腔镜模拟培训和考核研究现状分析与探讨