深度解析医学证据,DeepEvidence为你支撑决策
肺动脉高压是一类以肺动脉压力进行性升高和肺血管重构为特征的严重心肺血管疾病,起病隐匿但致死性高,最终可导致右心衰竭,严重威胁患者生命。缺氧是多种肺动脉高压发生发展的重要诱因之一,但缺氧状态下肺血管平滑肌细胞如何被持续激活、从而推动血管重构,其核心分子机制仍有待阐明。深入解析缺氧诱导肺动脉高压进展的调控网络,对于发现新的诊疗靶点、改善患者预后具有重要意义。
2026年5月26日,复旦大学基础医学院孟丹教授团队联合美国加州大学John Y-J. Shyy教授团队在《循环》(Circulation)杂志在线发表题为《缺氧介导BACH1表达上调并通过TGFBR2/SMAD信号通路加剧肺动脉高压》(“Hypoxia Upregulation of BACH1 Aggravates Pulmonary Hypertension Through TGFBR2/SMAD Pathways”)的研究论文。该研究发现,转录因子BACH1在缺氧条件下表达显著上调,并通过激活TGFBR2/SMAD信号通路,促进肺动脉平滑肌细胞表型转换及肺血管重构,从而加速肺动脉高压疾病的进展,提示BACH1是肺动脉高压治疗的潜在新靶点。

转录因子BACH1 (BTB and CNC homology 1)参与氧化应激、细胞周期调控、血红素稳态、肿瘤转移及免疫调控等多种生物过程。课题组前期研究已证实,血管内皮细胞BACH1缺失或BACH1小分子抑制剂可促进缺血后血管新生,内皮BACH1缺失对血管炎症和动脉粥样硬化具有保护作用(Circulation Research 2015, 2022;Molecular Therapy 2025);血管平滑肌细胞BACH1缺失可减少血管损伤后的小鼠新生内膜形成(Nucleic Acids Res. 2023)。然而,BACH1在肺动脉高压尤其是肺动脉平滑肌细胞中的作用及其上游/下游调控机制尚不清楚。研究团队首先结合特发性肺动脉高压病人(IPAH)样本和肺动脉高压动物模型发现,BACH1在肺动脉高压患者及动物的肺组织中明显升高,且主要富集于肺动脉平滑肌细胞中。
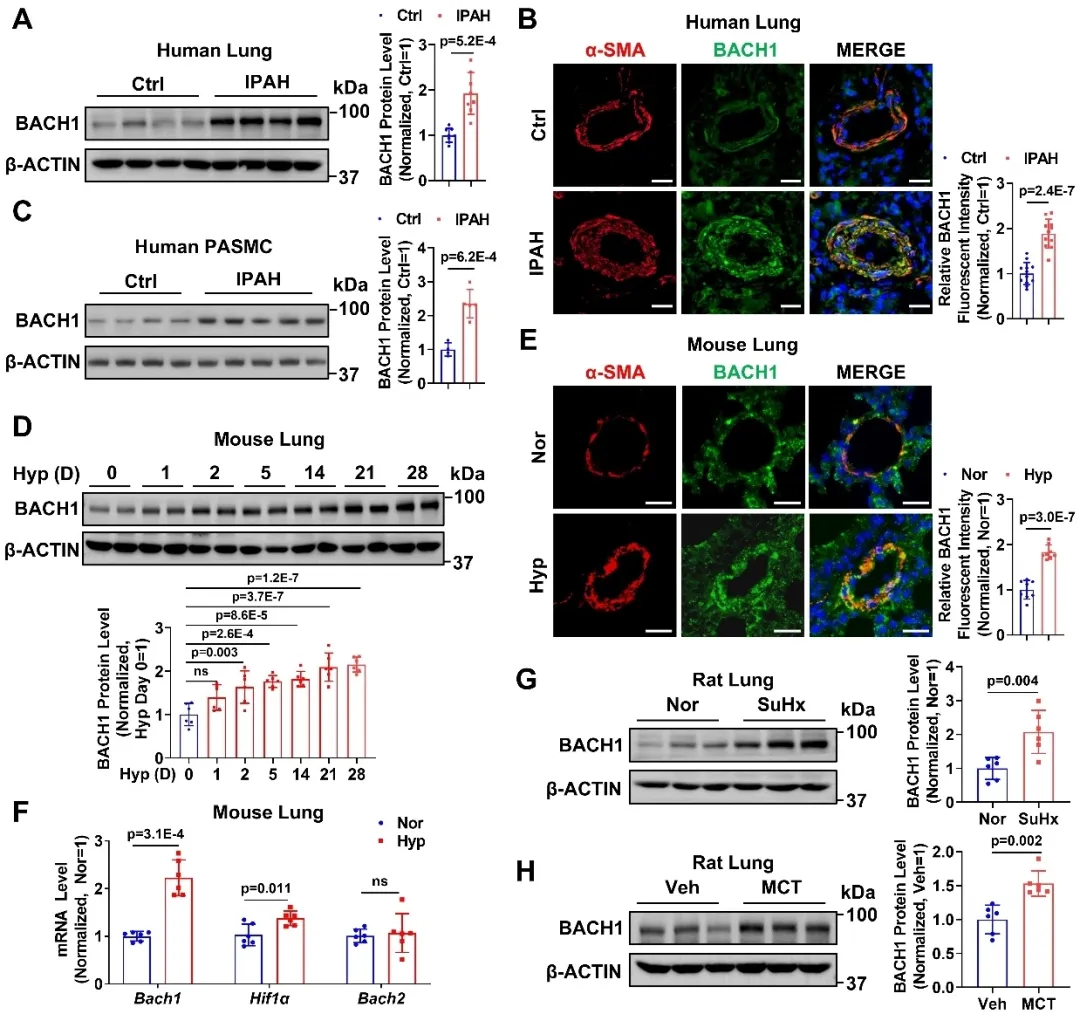
图1. BACH1在特发性肺动脉高压病人和肺动脉高压动物模型中表达上调
研究团队对缺氧条件下BACH1表达上调的分子机制进行探究,发现BACH1是脯氨酸羟化酶PHD2的底物,并且与E3泛素连接酶VHL存在结合。缺氧条件下,由于氧气不足,PHD2介导的BACH1羟基化修饰减少,导致BACH1与VHL的结合减弱,从而抑制泛素-蛋白酶体途径对BACH1的降解作用,增强BACH1的蛋白稳定性,并最终引起肺动脉平滑肌细胞中BACH1蛋白含量增多。以上研究结果揭示了BACH1通过氧浓度-PHD2信号轴实现蛋白稳态调控的新机制。
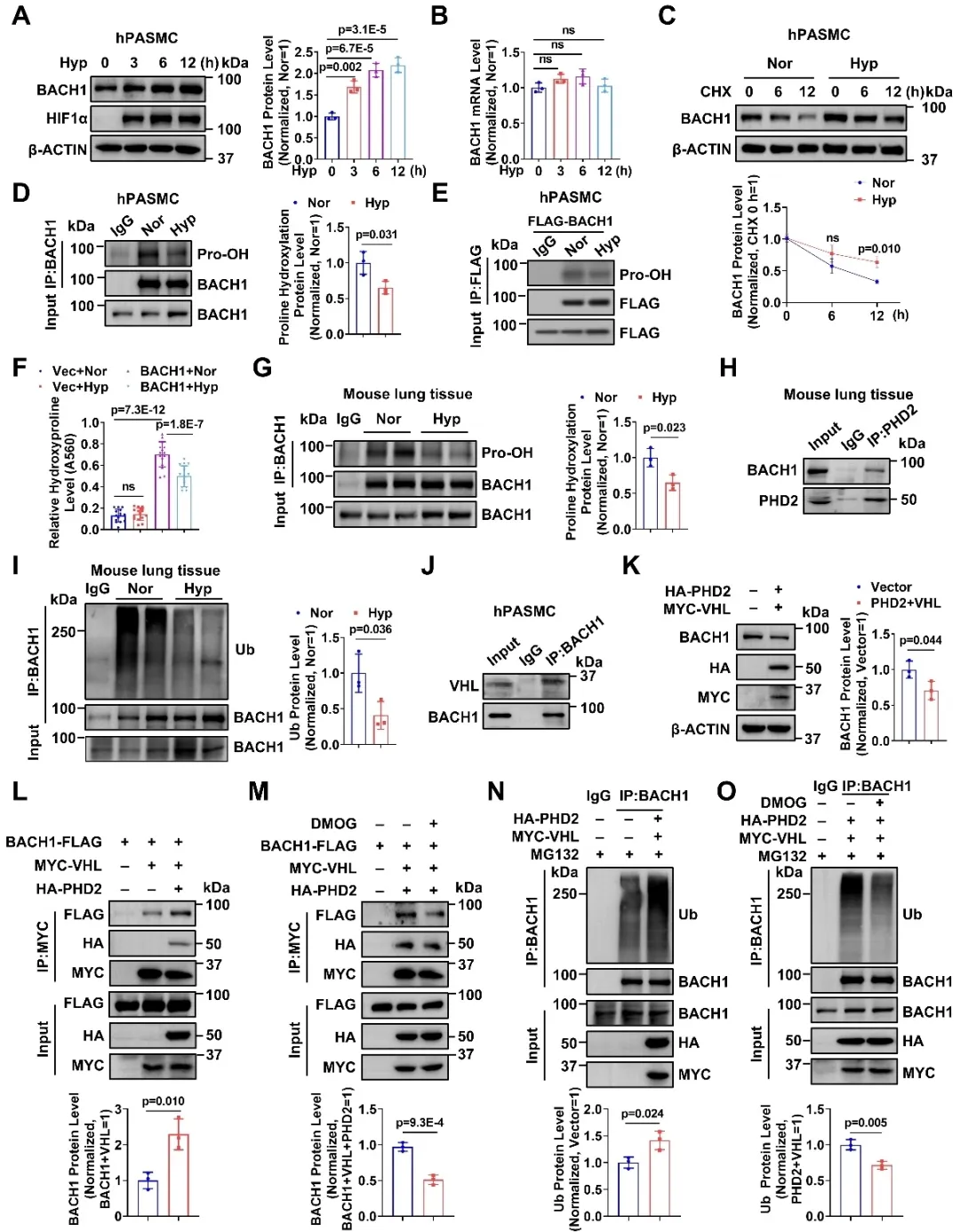
图2. BACH1以氧气/脯氨酸羟化酶依赖的方式发生脯氨酸羟基化修饰并被降解
为明确BACH1的功能,研究人员构建了血管平滑肌细胞特异Bach1基因敲除小鼠(Bach1SMCKO),并在缺氧条件下建立肺动脉高压小鼠模型。结果显示,与对照组相比,Bach1缺失可显著减轻肺动脉高压小鼠的右心室收缩压升高、右心室肥厚和肺动脉重构,改善肺血流动力学指标,以及减轻缺氧刺激造成的肺血管损伤。这些结果提示,BACH1在肺动脉高压发生发展中具有促进作用。
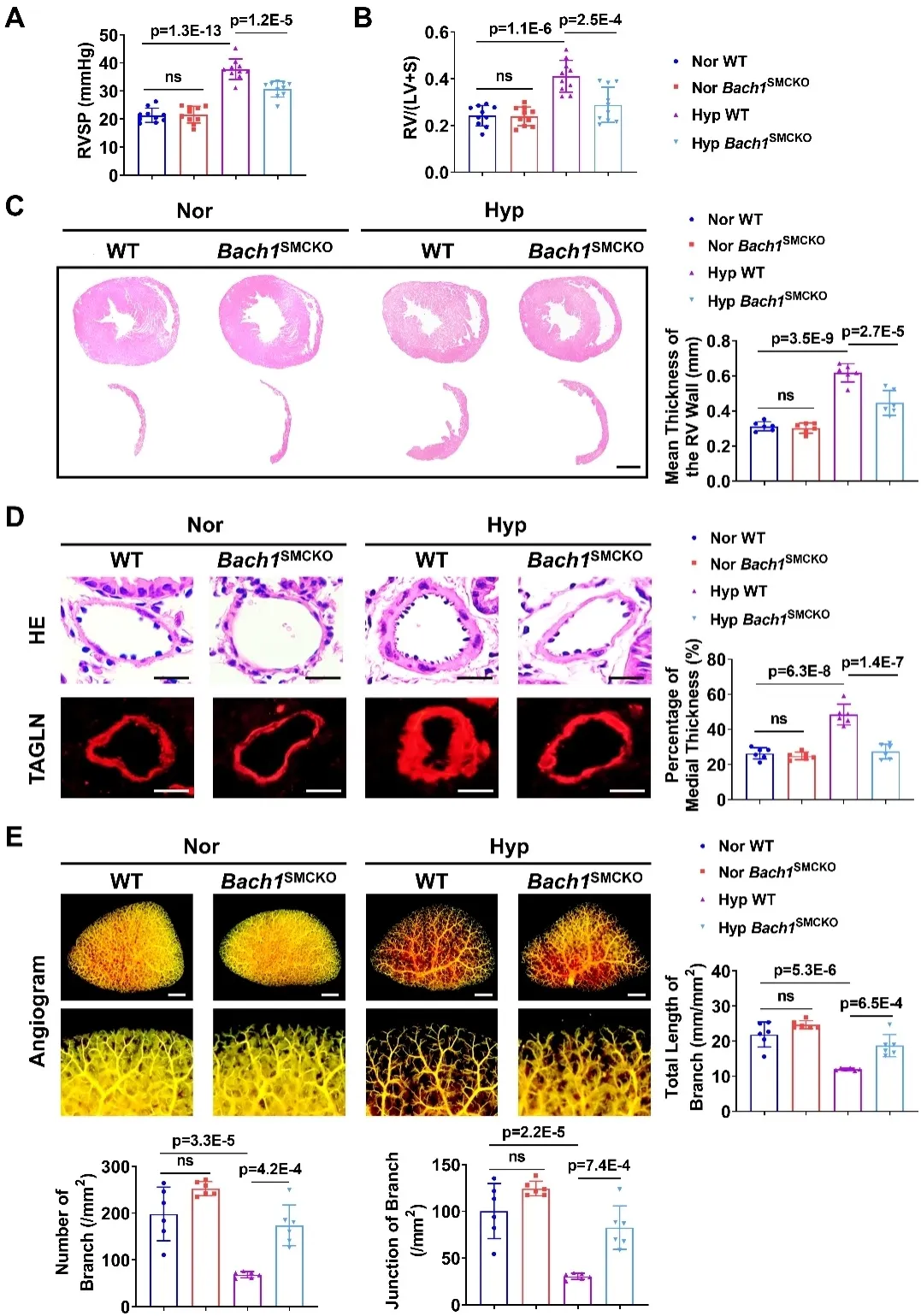
图3. 血管平滑肌细胞Bach1缺失减轻小鼠肺动脉高压表型
单核RNA测序分析进一步显示,平滑肌特异性Bach1敲除后,平滑肌细胞中SMAD3转录因子活性明显减弱,SMAD3是TGF-β通路的经典下游转录因子,提示BACH1可能通过TGF-β通路驱动病理性重构。TGFBR2作为TGF-β信号通路的重要受体,在血管重构、细胞增殖和细胞外基质沉积过程中发挥关键作用。机制研究显示,BACH1可直接结合TGFBR2启动子区域并促进其转录激活,从而上调TGFBR2表达并增强下游SMAD信号转导及细胞外基质基因的表达。动物体内验证发现,TGFBR2抑制剂处理可以缓解血管平滑肌Bach1基因过表达对小鼠肺动脉高压表型的促进作用。
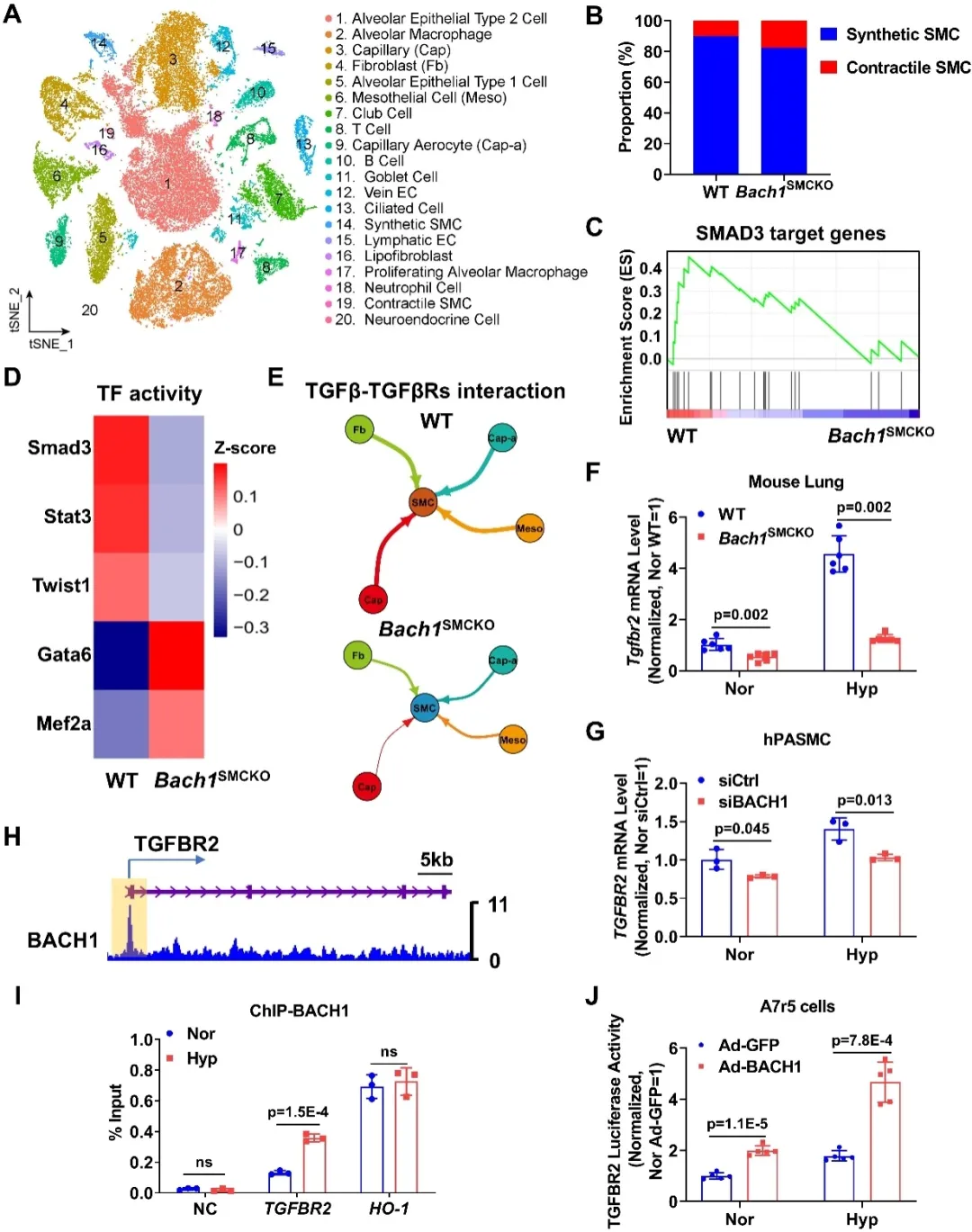
图4. BACH1结合在TGFBR2启动子区并激活其转录
为进一步验证BACH1在肺动脉高压疾病中对TGFBR2/SMAD信号通路的调控作用,研究人员向特发性肺动脉高压病人来源的肺动脉平滑肌细胞转染BACH1小干扰RNA。结果显示,BACH1在病人原代平滑肌细胞中表达升高,并且抑制BACH1可逆转病人原代细胞中原本异常激活的TGFBR2/SMAD信号通路,并下调细胞外基质相关基因的表达。上述结果共同证实,在肺动脉高压疾病中,BACH1与TGFBR2及细胞外基质沉积存在关联。
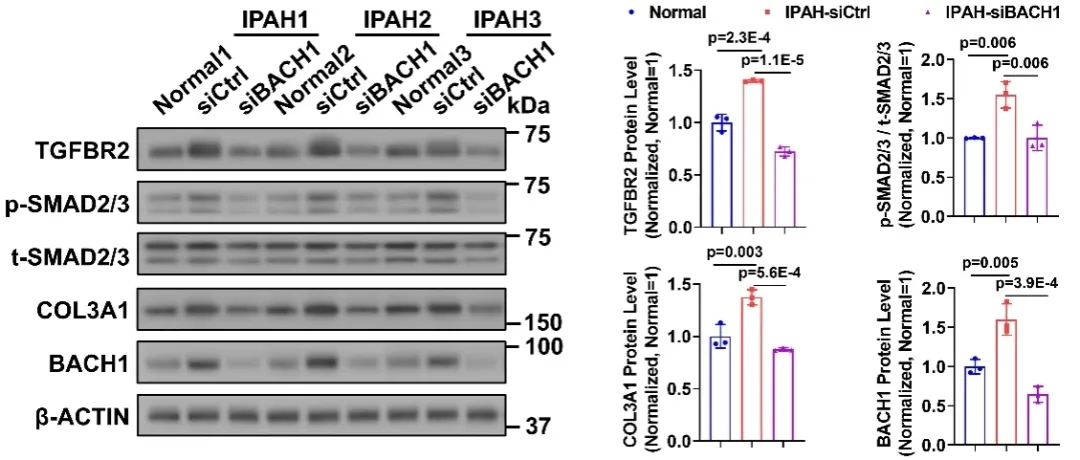
图5. BACH1抑制减弱IPAH-PASMCs中的TGFBR2/SMAD信号通路
综上,该研究系统阐明了“缺氧-BACH1-TGFBR2/SMAD”信号轴在肺动脉高压中的关键作用,揭示了BACH1作为缺氧应答转录因子,通过转录激活TGFBR2/SMAD信号,促进肺动脉高压进展。这一发现不仅深化了对肺动脉高压发病机制的认识,也为开发针对BACH1或TGFBR2/SMAD通路的新型治疗策略提供了潜在靶点。
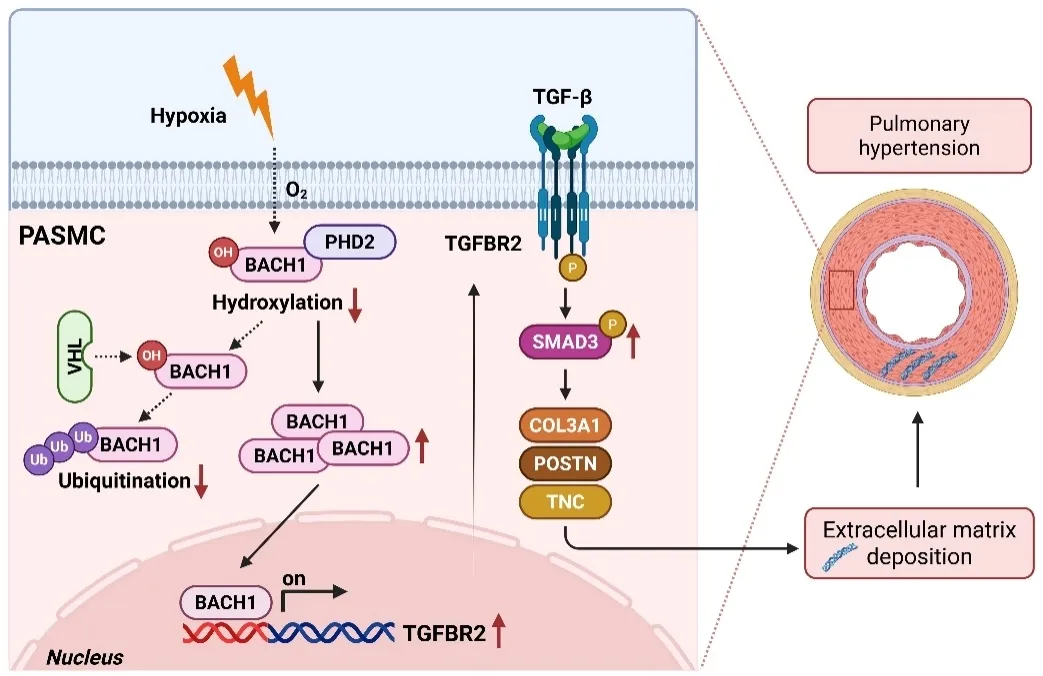
图6. 缺氧条件下BACH1表达上调并通过激活TGFBR2/SMAD通路加重肺动脉高压的机制模式图
复旦大学基础医学院孟丹教授、王新红副教授,美国加州大学圣地亚哥分校John Y-J. Shyy教授为论文的共同通讯作者,复旦大学基础医学院博士生侯燕楠、李沁函、美国加州大学圣地亚哥分校魏同佑(Wade)研究员、复旦大学基础医学院博士生林潇可及郭阶雨博士后为论文的共同第一作者,复旦大学为第一作者单位。该研究还得到了同济大学附属上海市肺科医院袁平主任医师、扬州大学附属医院沈慧主任医师、复旦大学附属中山医院徐晨主任医师和吴剑副研究员以及奥地利格拉茨医科大学Elena Osto教授的大力支持。该研究受到国家自然科学基金委、上海市科委以及中国博士后基金等项目的资助。
原文链接:
https://www.ahajournals.org/doi/10.1161/CIRCULATIONAHA.124.073606