深度解析医学证据,DeepEvidence为你支撑决策
心力衰竭是多种心血管疾病的终末阶段,而心肌能量代谢的重编程是其核心病理特征之一。在病理性心肌肥厚过程中,心肌细胞逐渐丧失“代谢灵活性”:脂肪酸氧化能力下降,糖酵解虽然代偿性增强,却与线粒体中的葡萄糖氧化发生“解偶联”,导致ATP生成速率下降,最终推动心功能持续恶化。然而,连接应激信号与葡萄糖氧化抑制的关键上游调控机制,长期以来尚未得到清晰阐明。
2026年5月22日,南方科技大学医学院覃刚健教授团队在心血管领域国际顶级期刊Circulation在线发表了题为“Sam68 Exacerbates Pathologic Cardiac Hypertrophy by Suppressing Cardiomyocyte Glucose Oxidation”的研究论文。研究首次鉴定RNA结合蛋白Sam68是压力超负荷诱导心肌代谢重塑的关键驱动因子,并系统揭示其通过“Sam68–STAT3–PDK4”信号轴抑制丙酮酸脱氢酶(PDH)活性、阻断葡萄糖氧化的全新机制,为心力衰竭的代谢干预提供了潜在可成药的靶点。

研究团队首先通过挖掘扩张型心肌病、缺血性心肌病及肥厚型心肌病等公共转录组数据发现,Sam68在衰竭心脏中显著上调,且主要定位于心肌细胞。在血管紧张素II (Ang II)输注及主动脉缩窄(TAC)小鼠模型中,这一发现得到进一步验证:压力应激可迅速诱导Sam68心肌细胞核内积聚。
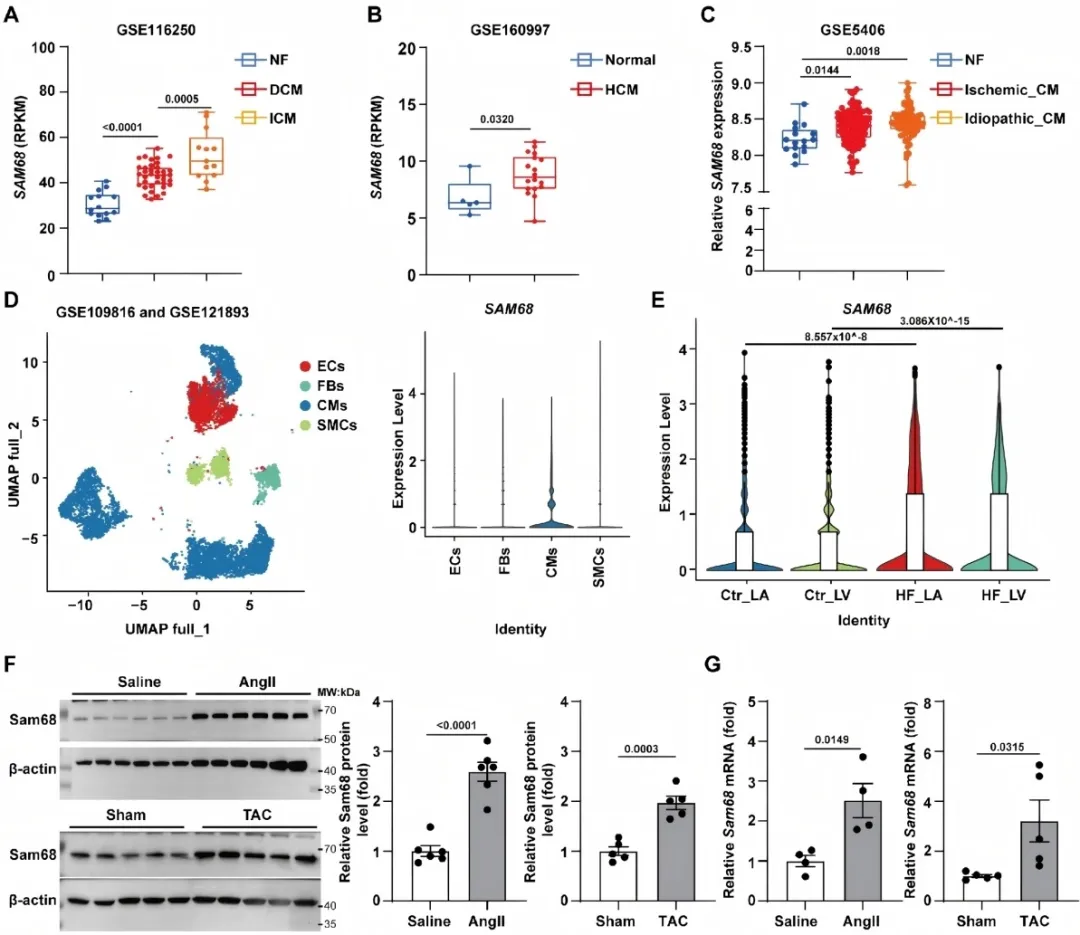
图1. Sam68在人类衰竭心脏及小鼠压力超负荷肥厚模型中表达上调
功能研究进一步证实了Sam68在病理性心脏重构中的关键作用。研究人员构建心肌细胞特异性Sam68敲除小鼠后发现,无论是TAC还是Ang II诱导的病理性肥厚、心腔扩张及心肌纤维化均显著减轻,心功能得到明显保护;相反,通过AAV9在心肌细胞中特异性过表达Sam68,则显著加重压力超负荷诱导的心脏重构及收缩功能障碍。
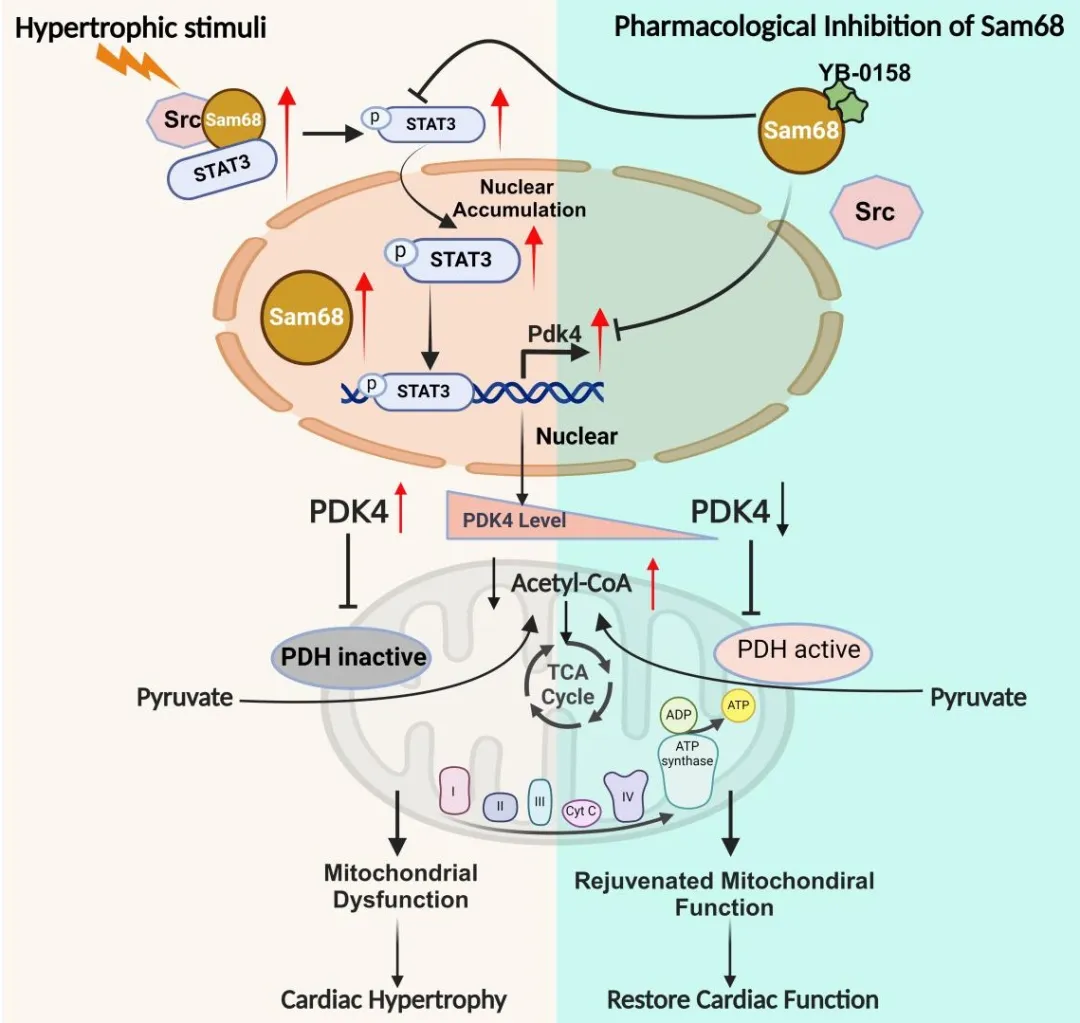
为深入解析Sam68加重心肌肥厚的调控机制,研究团队整合了RNA-seq、靶向代谢组学及体内同位素示踪技术。结果显示,压力超负荷导致心肌细胞内糖酵解中间产物堆积,但葡萄糖碳进入三羧酸(TCA)循环却显著减少,呈现典型的“糖酵解-氧化解偶联”。而Sam68敲除则能逆转这一代谢障碍:恢复PDH依赖的丙酮酸氧化,使葡萄糖碳顺利进入TCA循环,从而改善心肌能量代谢。进一步的机制研究揭示,Sam68在此过程中并非依赖其经典的RNA剪接功能,而是作为一种“信号支架蛋白”发挥功能。在应激条件下,Sam68与Src家族激酶结合,招募并促进STAT3的Tyr705位点磷酸化及核转位;入核后的STAT3进一步转录激活丙酮酸脱氢酶激酶4 (PDK4),随后PDK4磷酸化PDH使其失活,从而阻断糖酵解与线粒体氧化的代谢衔接。由此,研究团队勾勒出一条清晰的“Sam68→Src→STAT3→PDK4→PDH” 病理信号轴。该研究建立了“应激信号—转录调控—代谢抑制”之间的直接联系,为理解心衰代谢重塑提供了新的理论框架。
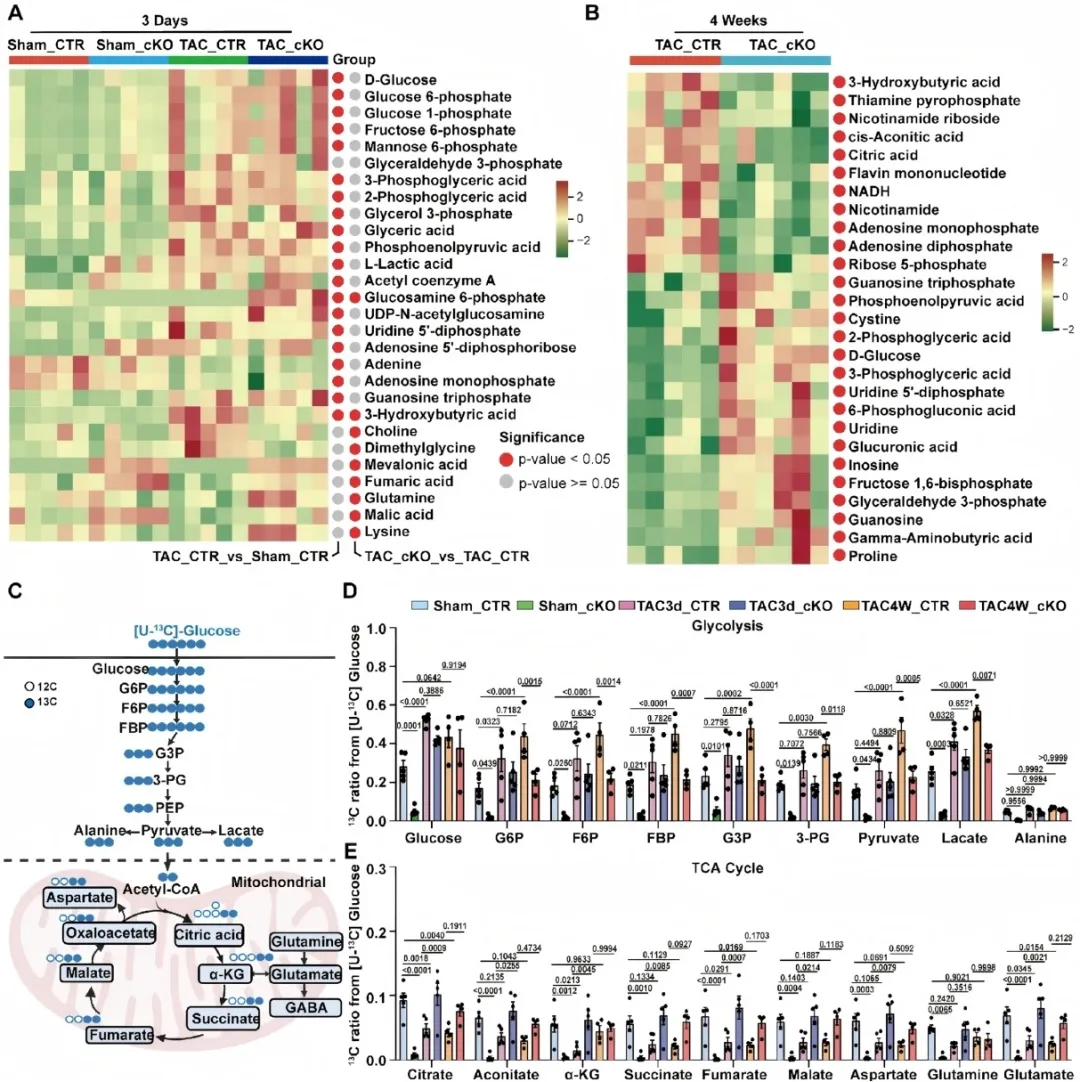
图2. Sam68缺失促进压力超负荷下丙酮酸氧化及葡萄糖碳进入三羧酸循环
在干预层面,研究团队验证了该通路的治疗潜力。PDK4选择性抑制剂能够显著缓解Sam68过表达小鼠的心肌肥厚,恢复PDH活性并改善线粒体功能。更进一步,团队验证了一种靶向Sam68–Src蛋白互作的小分子抑制剂YB-0158。该化合物在心脏组织中具有良好的暴露水平和靶点占有率,可有效破坏Sam68与Src的结合并抑制下游信号激活。在TAC诱导的压力超负荷模型中,YB-0158显著改善心功能并减轻心脏重构。而这一保护效应在Sam68敲除小鼠中完全消失,严格证明了其作用依赖于Sam68靶向机制。
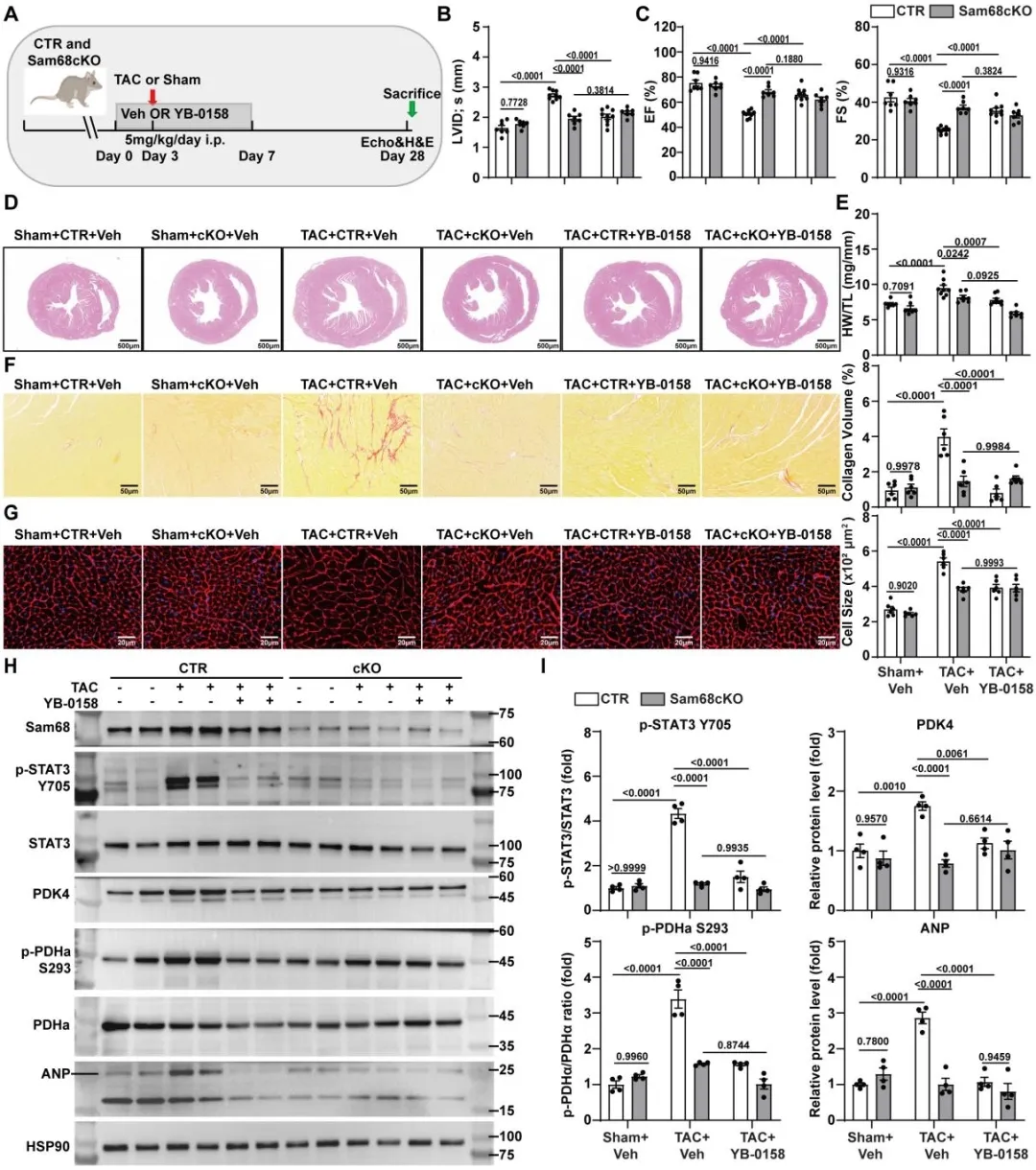
图3. 靶向阻断Src–Sam68相互作用以Sam68依赖方式减轻TAC诱导的心脏重构
为评估该机制在人类疾病中的临床意义,研究团队进一步分析了终末期心衰患者左心室组织。结果显示,p-Src、Sam68、p-STAT3 (Y705)、PDK4及p-PDHα水平均较非衰竭心脏显著升高。同时,Sam68蛋白表达与PDK4及心衰标志物ANP呈正相关趋势,与左室射血分数(LVEF)呈负相关。这表明,Src–Sam68–STAT3–PDK4信号轴在人类衰竭心脏中同样处于显著激活状态。
综上,该研究揭示了一条全新的心力衰竭代谢重塑机制:在压力超负荷等病理应激下,心肌细胞中的Sam68被激活,并作为信号支架驱动STAT3核转位及PDK4转录激活,最终持续抑制PDH活性、阻断葡萄糖氧化,加速病理性心肌肥厚及心衰进展。该研究不仅深化了对心衰代谢重编程机制的认识,也为代谢性心力衰竭的精准干预提供了新的理论依据。靶向Sam68 (尤其是阻断Sam68–Src蛋白互作)或选择性抑制PDK4以恢复心肌葡萄糖氧化效率,有望成为未来心衰治疗的重要新策略。
南方科技大学医学院覃刚健教授为本文通讯作者,研究助理教授安俊卿、韩超珊为共同第一作者。该研究由覃刚健团队联合华中科技大学同济医学院附属协和医院董念国教授、重庆大学附属重庆市人民医院吕青研究员、南方科技大学医学院冯宇亮教授及曹阳坡教授共同完成,并获得了国家自然科学基金、中国博士后科学基金、深圳市医学研究基金及广东省自然科学基金等项目的资助。
原文链接:
https://www.ahajournals.org/doi/10.1161/CIRCULATIONAHA.125.077533