深度解析医学证据,DeepEvidence为你支撑决策
在错配修复蛋白缺陷或微卫星不稳定(MSI)的结直肠癌患者中,高达 50% 的病例不存在MMR基因胚系突变、BRAF突变或MLH1启动子甲基化。这类病例被定义为林奇样综合征(LLS)。林奇样综合征是一组异质性疾病,可能包含所有表现为微卫星不稳定、属于林奇综合征肿瘤谱,但未检测到MMR基因致病变异的患者。尽管目前已提出多种方法用于区分林奇综合征和林奇样综合征,但针对这类患者的精准分类仍缺乏共识。本文报道了日常临床病理诊断工作中遇到的 4 例疑似林奇样综合征病例。分析了相关肿瘤的组织病理学特征和分子病理学改变,并通过文献回顾梳理了该疾病的诊疗进展。结合临床表现和分子病理学检测结果,2 例被诊断为林奇样综合征(LLS),2 例被诊断为非典型表型林奇综合征。林奇样综合征相关肿瘤可发生于结直肠和肠外器官。结直肠肿瘤无特定的发病部位或组织学特征,而肠外肿瘤常表现为低分化,伴大量间质淋巴细胞浸润。所有林奇样综合征患者的肿瘤病灶均存在错配修复蛋白(MLH1、PMS2、MSH2、MSH6)表达缺失、微卫星不稳定(MSI-L/MSI-H)、BRAF野生型以及MLH1启动子甲基化阴性的特征。但同一患者不同部位的肿瘤之间,在MMR蛋白表达、MSI状态和MLH1启动子甲基化情况上存在异质性。所有林奇样综合征病例均未检测到MMR基因胚系致病变异,但 1 例存在MMR基因意义不明的变异,2 例(非典型表型林奇综合征)存在MLH1基因疑似致病变异。林奇样综合征相关的肠外肿瘤大多表现出与经典林奇综合征相似的组织病理学特征及MMR/MSI改变,但不存在MMR基因胚系致病变异或MLH1启动子甲基化。部分存在MMR基因胚系疑似致病变异的疑似林奇样综合征病例,可能属于非典型表型的林奇综合征。大多数病例的正常组织中不存在MMR基因胚系突变,但肿瘤组织中存在MMR基因体细胞突变。部分病例可检测到其他与MMR功能相关基因的胚系或体细胞突变。
背 景
林奇综合征(LS)由错配修复(MMR)基因的胚系突变所致,会引发DNA修复功能障碍,以家族聚集性和早发性为特征。准确诊断对于家族筛查和临床干预至关重要。但部分患者表现出与林奇综合征相似的临床特征(如早发疾病、多原发癌或相关家族史),却检测不到可识别的MMR基因胚系突变,这类病例被称为“林奇样综合征”。目前该领域仍面临诸多挑战:定义尚未统一,诊断标准缺乏共识,针对病理特征的系统性研究也存在不足。现有研究大多基于小样本,缺乏对临床病理特征的全面总结。因此,本研究回顾性分析了 4 例临床疑似林奇样综合征病例的临床资料、病理特征及免疫组化表型,结合文献综述,探讨其临床病理特征,为该疾病的鉴别诊断和临床管理提供参考。
研究材料与方法
案例收集
本研究纳入 2024 年 1 月至 10 月期间在中国人民解放军火箭军特色医学中心病理科确诊的 4 例疑似林奇样综合征病例。所有患者的肿瘤病灶均具备以下特征:错配修复蛋白(MLH1、PMS2、MSH2、MSH6)表达缺失、微卫星不稳定(MSI-L/MSI-H)、BRAF基因野生型以及MLH1启动子甲基化阴性。所有病例均经两名副主任医师及以上职称的病理医师复阅确认,本研究已获得机构伦理委员会批准,且所有患者均已签署书面知情同意书。
错配修复蛋白的免疫组化检测
免疫组化染色采用全自动染色平台,运用EnVision两步法完成。判读标准如下:MLH1、PMS2、MSH2、MSH6的阳性信号定位于细胞核。肿瘤细胞染色阳性表现为细胞核呈棕褐色,邻近的正常上皮细胞或间质细胞作为内阳性对照,细胞核同样呈棕褐色。若肿瘤细胞无细胞核染色,但周边正常细胞可见棕褐色细胞核染色,则判定为肿瘤细胞染色阴性。
MLH1启动子甲基化分析
从肿瘤组织中提取DNA,使用紫外分光光度计测定其浓度和纯度。按照生产商说明书,完成亚硫酸氢盐修饰及修饰后DNA的纯化。在荧光定量仪上通过实时定量PCR检测MLH1甲基化状态,以COLO2A1作为内参。
MMR基因的胚系和体细胞分析
收集患者外周血样本及石蜡包埋的原发/转移肿瘤组织。采用下一代测序(NGS)全外显子测序技术检测MMR基因的胚系突变和体细胞突变,同时评估肿瘤的微卫星不稳定(MSI)状态。检测基因列表包含 22 个与遗传性消化系统肿瘤相关的基因,即APC、ATM、AXIN2、BLM、BMPR1A、CHEK2、EPCAM、GALNT12、GERM1、MLH1、MSH2、MSH3、MSH6、MUTYH、NTHL1、PMS2、POLD1、POLE、PTEN、SMAD、STK11、TP53。依据美国医学遗传学与基因组学学会/美国分子病理学学会(ACMG/AMP)序列变异解读标准与指南,通过对“致病证据”和“良性证据”进行“加权评估”,最终将变异分为 5 类:致病性、疑似致病性、意义不明确、疑似良性、良性。
肿瘤组织中微卫星不稳定性检测
微卫星不稳定(MSI)采用2B3D荧光定量PCR-毛细管电泳法进行检测,所用标志物包括BAT-25、BAT26、D2S123、D17S250和D5S346。判定标准:≥2 个标志物存在不稳定定义为微卫星高度不稳定(MSI-H),1 个标志物存在不稳定定义为微卫星低度不稳定(MSI-L),所有标志物均稳定则定义为微卫星稳定(MSS)。
研究结果
临床特征
病例1:患者男性,53 岁,因体检发现直肠占位性病变 2 天就诊。电子结肠镜检查提示直肠距肛缘 13-16 cm处可见环周黏膜隆起,伴肠腔狭窄。患者接受了腹腔镜下直肠癌前切除术、腹腔淋巴结清扫术及肠粘连松解术。
病例2:患者女性,62 岁,因绝经后间歇性阴道流血 2 年、近 4 个月症状加重入院。PET-CT显示宫腔及降结肠部位FDG摄取增高,可疑恶性肿瘤。
病例3:患者女性,37 岁,因月经紊乱1年、间期阴道排液 2 个月就诊。盆腔超声提示宫颈前唇处可见不均质回声(大小 2.8×2.8×2.0 cm,边界不清、形态不规则),盆腔MRI证实为 2.9×2.1 cm的肿块。乳腺超声提示左侧乳腺低回声结节(大小 15×10×13 mm,BI-RADS 4a类)。患者接受了子宫内膜癌全面分期手术,以及左乳保乳根治术联合前哨淋巴结活检术。
病例4:患者女性,43 岁,因体检发现结肠多发息肉 3 天入院。结肠镜检查提示距肛缘40-80 cm处可见数十枚息肉样隆起(大小 0.2-2.5 cm),最大病灶为 2.5×1.2 cm(带蒂)。结肠镜初步诊断为“结肠多发息肉,性质待查”,临床可疑家族性腺瘤性息肉病。患者接受了腹腔镜下全结肠切除术+回肠直肠吻合术。
宏观和微观结果
病例1:距远端切缘 3 cm处可见一环周溃疡性肿块,大小为 6×5×1 cm,中心坏死凹陷。镜下观察可见肿瘤细胞形成腺管样结构,侵透肠壁全层。肿瘤间质可见密集淋巴细胞浸润,浸润边缘可见三级淋巴滤泡形成。病理诊断为“直肠中分化管状腺癌”。
病例2:送检标本包含一段结肠、全子宫及双侧附件。结肠肿瘤:呈腺管样结构,局灶筛状融合,侵透肠壁。诊断为“结肠中分化腺癌”。子宫内膜肿瘤:呈实性巢状浸润肌层,可见血管内癌栓。诊断为“子宫内膜低分化子宫内膜样腺癌”。
病例3:送检标本包含全子宫、双侧附件及左乳肿瘤。子宫肿瘤:呈腺管样及实性巢状结构,局灶可见角化珠,巢间密集淋巴细胞浸润,浸润宫体及宫颈。诊断为“中-低分化子宫内膜样腺癌,局灶伴腺鳞癌特征”。乳腺肿瘤:圆形/多边形细胞呈实性巢状排列,伴丰富淋巴细胞浸润。诊断为“伴髓样特征的浸润性乳腺癌”。
病例4:切除结肠长 55 cm,直径 3-7 cm,距近端切缘 5 cm处可见长 6 cm的阑尾。肠壁散在 15 枚息肉(粟粒至米粒大小),其余黏膜正常。肿瘤细胞呈腺管样结构,核异型性轻微。诊断为“结肠低级别腺瘤性息肉”(图1)。
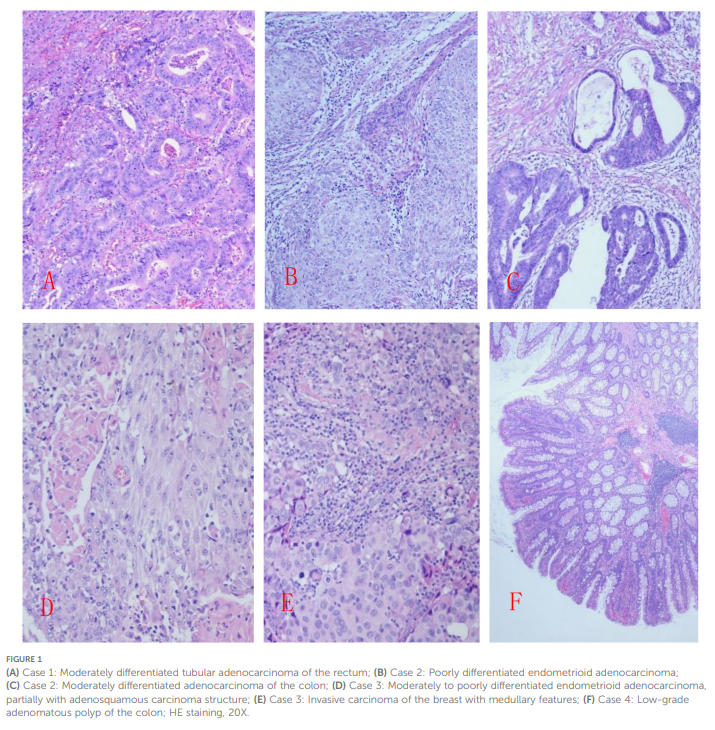
▲图1
(A)病例 1:直肠中分化管状腺癌;(B)病例 2:低分化子宫内膜样腺癌;(C)病例 2:结肠中分化腺癌;(D)病例 3:中低分化子宫内膜样腺癌,部分伴有腺鳞癌结构;(E)病例 3:乳腺浸润性癌,伴有髓样特征;(F)病例 4:结肠低级别腺瘤性息肉;HE 染色,20 倍
免疫表型
病例1:肿瘤细胞CK20、CDX2、MLH1、PMS2表达阳性,MSH2、MSH6染色阴性。
病例2:子宫内膜肿瘤细胞CK7、ER、PR、MLH1、PMS2阳性,CK5/6、P63、P40、PTEN、PAX2、MSH2、MSH6阴性;结肠肿瘤细胞CK20、CDX2、MLH1、PMS2、MSH2、MSH6阳性,CK7阴性。
病例3:宫颈肿瘤细胞CK7、CK5/6、P63、P40、ER、PR、MSH2、MSH6阳性,PTEN、PAX2、MLH1、PMS2阴性;乳腺肿瘤细胞CK7、E-钙黏蛋白、GATA3、ER、PR、HER2、MSH2、MSH6阳性,CK5/6、P63、P40、MLH1、PMS2阴性。
病例4:肿瘤细胞CK20、CDX2、MSH2、MSH6阳性,MLH1、PMS2表达阴性(图2)。
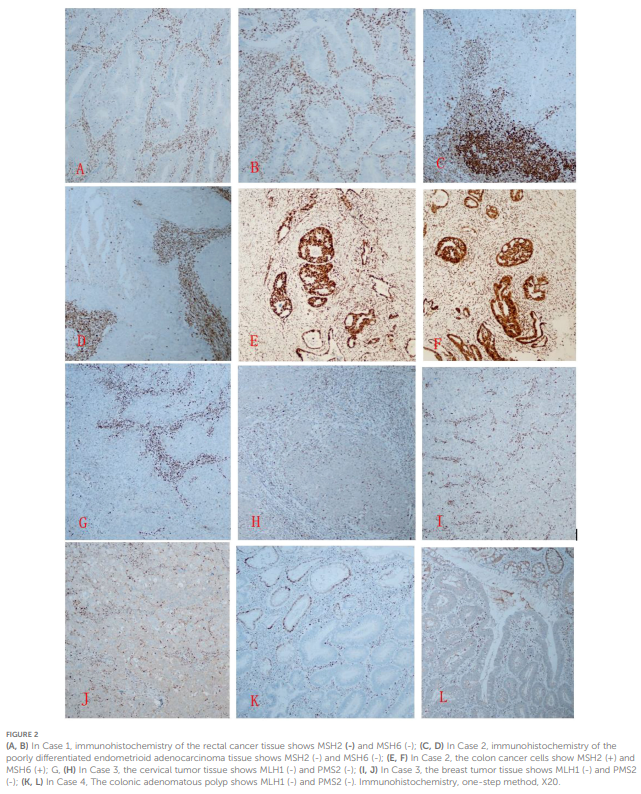
▲图2
(A、B)病例1中,直肠癌组织免疫组化显示MSH2 (-)和MSH6(-);(C、D)病例2中,低分化子宫内膜样腺癌组织免疫组化显示MSH2(-)和MSH6(-);(E、F)病例2中,结肠癌细胞显示MSH2(+)和MSH6(+);(G、H)病例3中,宫颈肿瘤组织显示MLH1(-)和PMS2(-);(I、J)病例3中,乳腺肿瘤组织显示MLH1(-)和PMS2(-);(K、L)病例4中,结肠腺瘤性息肉显示MLH1(-)和PMS2(-)
分子病理特征
病例1:外周血胚系突变分析显示PMS2基因存在意义不明的变异(VUS)。直肠癌组织下一代测序(NGS)检测到MSH2、MSH6基因存在体细胞致病性突变,PMS2基因存在意义不明的变异(VUS)。肿瘤为微卫星高度不稳定(MSI-H),无BRAF基因突变,MLH1启动子甲基化阴性。
病例2:外周血未检测到MMR基因胚系突变。子宫内膜癌组织NGS检测到POLE基因致病性突变,结肠癌组织检测到TP53基因致病性突变,未发现MMR基因改变。子宫内膜癌为微卫星低度不稳定(MSI-L),结肠癌为微卫星稳定(MSS)。
病例3:外周血、子宫及乳腺肿瘤组织中均检测到MLH1基因疑似致病性突变。肿瘤组织为微卫星高度不稳定(MSI-H),无BRAF基因突变,MLH1启动子甲基化阴性。
病例4:外周血胚系分析显示MLH1基因存在疑似致病性突变。结肠肿瘤组织NGS检测到TP53基因致病性突变及MLH1基因疑似致病性突变。肿瘤为微卫星高度不稳定(MSI-H),无BRAF基因突变,MLH1启动子甲基化阴性(表1、表2)。
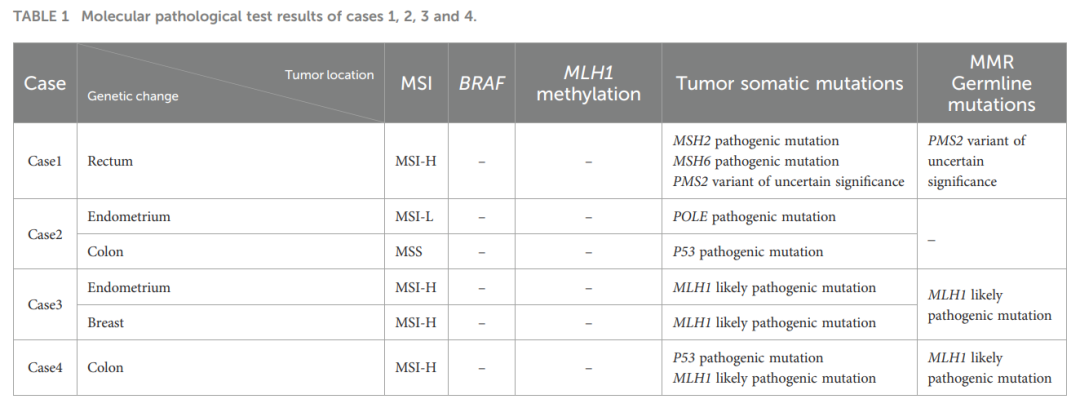
▲表1 病例1、2、3和4的分子病理学检测结果
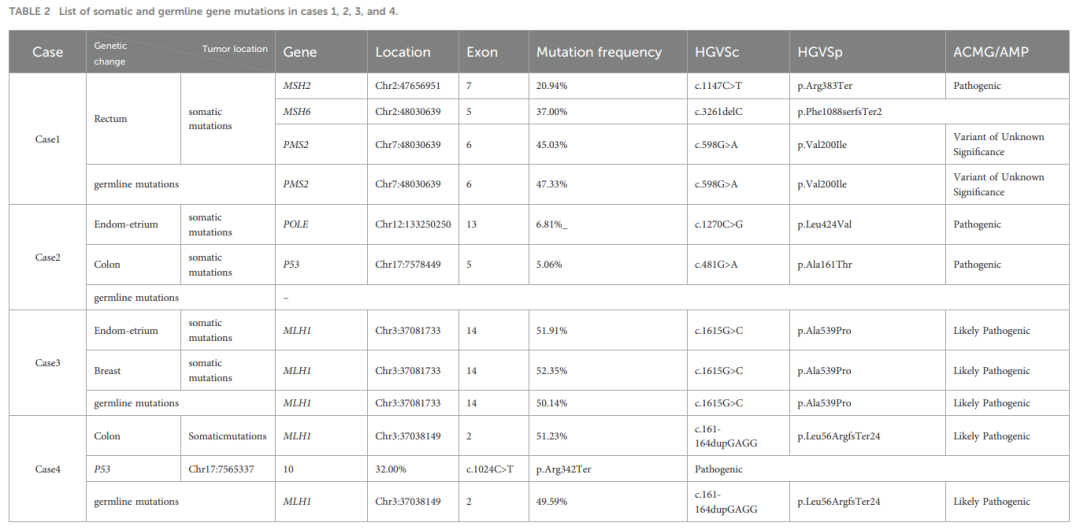
▲表2 病例1、2、3和4中的体细胞和胚系基因突变列表
讨 论
林奇样综合征(LLS)是指具有与林奇综合征相似的临床表型,但检测不到错配修复基因(MMR)胚系致病性/疑似致病性突变的一类异质性疾病。尽管目前已提出多种方法用于区分林奇样综合征的遗传性和散发病例,但针对这类患者的精准分类仍缺乏共识。关于林奇样综合征(LLS)的定义,部分学者认为其应指具有遗传性癌症可疑特征(如早发疾病、多原发癌或相关家族史),但未检测到MMR基因致病变异的病例。根据这一定义,仅存在微卫星不稳定(MSI)的癌症不足以将患者归类为林奇样综合征,还需满足阿姆斯特丹标准和/或贝塞斯达标准。另一种定义以Picó等学者为代表,将所有表现为肿瘤微卫星不稳定(MSI)或MMR蛋白表达缺失,但无MMR基因胚系致病变异证据的患者均纳入林奇样综合征范畴,这是一种更宽泛的定义,因为所有存在MSI且无MMR基因突变的结直肠癌/子宫内膜癌患者都被包含在内。依据国际分类准则妥善处理意义不明的变异(VUS)十分重要,MMR基因中分类不明确的VUS也可能是林奇样综合征的致病原因。本文基于林奇样综合征(LLS)的扩展定义,选取 4 例病理疑似林奇样综合征的临床病例,分析相关肿瘤的组织病理学特征和分子病理学改变。结合临床表现和分子病理学检测结果,2 例被诊断为林奇样综合征(LLS),2 例被诊断为非典型表型林奇综合征。
病例1的大体和镜下表现均符合经典直肠中分化管状腺癌,但免疫组化检测显示较为罕见的MSH2/MSH6表达缺失,微卫星不稳定(MSI)检测证实肿瘤为微卫星高度不稳定(MSI-H)。MLH1启动子甲基化和BRAF基因突变检测结果不支持经典散发性错配修复缺陷(dMMR)结直肠癌的诊断。由于MMR胚系突变分析仅检测到PMS2基因的意义不明变异(VUS),故排除林奇综合征。结合肿瘤组织中检测到MSH2和MSH6基因的体细胞致病性突变,该病例被诊断为林奇样综合征。临床实践中,经典散发性MSI-H结直肠癌主要由MLH1启动子甲基化所致,且常伴随BRAF基因突变。由MSH2或MSH6基因胚系致病性突变导致的林奇综合征通常表现为同时性或异时性肠外恶性肿瘤,且发病年龄更高。尽管该病例检测到PMS2基因的胚系VUS,但其临床意义尚不明确,不能作为诊断林奇综合征的依据。
病例2患者同时患有低分化子宫内膜样腺癌和结肠中分化管状腺癌。子宫内膜肿瘤组织显示MSH2/MSH6蛋白表达缺失,且为微卫星低度不稳定(MSI-L)。尽管患者肿瘤发病年龄相对较高,但临床和病理特征高度提示林奇综合征。进一步评估显示,结肠癌的MSH2/MSH6表达保留,且为微卫星稳定(MSS),缺乏林奇综合征相关肿瘤的分子病理学特征。外周血MMR基因胚系突变分析结果为阴性,排除林奇综合征。肿瘤组织NGS测序检测到子宫内膜癌中存在POLE基因致病性突变,结肠癌中存在P53致病性突变。鉴于POLE基因突变可诱导MMR蛋白表达缺失,该病例被诊断为POLE致病性突变所致的林奇样综合征。结直肠癌和子宫内膜癌是最常见的林奇相关肿瘤,尽管该患者同时患有子宫内膜癌和结肠癌,但两种肿瘤的MMR蛋白表达和微卫星状态存在显著差异,且未检测到MMR基因胚系突变,这与林奇综合征的分子特征不符。POLE体细胞突变在子宫内膜癌中较为常见,已有研究报道POLE突变可导致MMR蛋白表达缺失和微卫星不稳定,与本病例的发现一致。低分化子宫内膜样腺癌相对少见,存在MMR蛋白缺失的肿瘤常表现为低分化和髓样癌表型,伴丰富的淋巴细胞间质,且预后优于常规低分化腺癌。
病例3为年轻女性,同时患有子宫内膜样腺癌(局灶伴腺鳞分化)和乳腺浸润性导管癌。两种肿瘤均表现为MLH1/PMS2蛋白表达缺失和微卫星高度不稳定(MSI-H),未检测到BRAF突变或MLH1启动子甲基化。通过外周血NGS测序检测到MLH1基因疑似致病变异,因此诊断为非典型表型林奇综合征。研究表明,部分遗传性病例可能是由罕见MMR突变导致的经典林奇综合征。尽管患者确诊时未发生结直肠癌,但其父亲在 50 岁前因胃癌、结直肠癌和膀胱癌去世,因此不能完全排除由罕见MMR突变导致的林奇综合征可能。应结合基因功能、变异特征、临床资料及其他相关信息进行综合评估,以排除非典型表型的林奇综合征。乳腺癌和子宫内膜癌是常见的遗传相关恶性肿瘤,前者主要与BRCA1/2胚系突变导致的遗传性乳腺癌卵巢癌综合征(HBOC)相关,后者则与MMR基因突变导致的林奇综合征密切相关。乳腺癌是林奇综合征较为少见的肠外表现。本病例中,子宫内膜肿瘤和乳腺肿瘤均表现为MLH1/PMS2表达缺失、MSI-H,且低分化区域可见明显淋巴细胞浸润,这些组织学特征均与林奇相关肿瘤相符。该病例曾因外周血MMR胚系突变检测结果被误报为意义不明的变异(VUS)而被误诊为林奇样综合征,后在评审专家的指导下纠正了诊断。这进一步证实,对于部分有明确家族史且存在MMR胚系突变的病例,诊断林奇样综合征时应极其谨慎。
病例4患者在体检中发现结肠多发息肉,临床初步诊断为“家族性腺瘤性息肉病(FAP)”。镜下观察可见腺体呈低级别异型增生,病理诊断为“低级别腺瘤性息肉”。肿瘤组织表现为MLH1/PMS2蛋白表达缺失和微卫星高度不稳定(MSI-H),未检测到BRAF突变或MLH1启动子甲基化。外周血胚系突变分析检测到MLH1基因疑似致病变异,未检测到APC、MUTYH、NTHL1等FAP/MAP相关基因的胚系致病性突变。患者父亲临床诊断为家族性腺瘤性息肉病(FAP),但尚未完成遗传性消化系统肿瘤相关基因的胚系突变检测,因此该病例病理诊断为非典型表型林奇综合征。FAP是一种罕见的常染色体显性遗传病,以多发腺瘤性息肉为特征,易导致早发性结直肠癌(CRC)。约 70% 的FAP患者存在肠外表现,如加德纳综合征、特科特综合征或胃腺癌伴近端胃息肉病(GAPPS),这些均与腺瘤性息肉病coli(APC)基因的胚系突变相关。APC基因突变是经典型FAP(cFAP)的主要病因,但未检测到APC突变的APC(-)/cFAP病例通常存在MUTYH、NTHL1等易感基因的胚系突变。双等位基因错配修复(MMR)突变可导致常染色体隐性遗传的APC(-)/cFAP,而常染色体显性遗传型则可能由POLE/POLD1、AXIN2或DUOX2基因突变引起。尽管本病例最终诊断为非典型表型林奇综合征,但其对林奇样综合征的鉴别诊断具有参考意义:对于同样存在错配修复蛋白表达缺失和微卫星不稳定表型的病例,若检测到MMR基因的意义不明变异(VUS),应诊断为林奇样综合征。
研究表明,林奇样综合征患者及其一级亲属患结直肠癌和其他林奇综合征(LS)相关癌症的风险低于林奇综合征患者,但林奇样综合征患者的结直肠癌风险仍高于散发病例。María Dolores Picó等人对 160 例林奇样综合征患者进行分析,结果显示林奇样综合征相关结直肠癌的平均发病年龄为 55 岁,其中女性占 41%;11% 的患者符合林奇综合征的阿姆斯特丹I/II标准,65% 符合修订版贝塞斯达指南;24% 的林奇样综合征患者是在结直肠癌筛查中被发现的。在符合阿姆斯特丹/贝塞斯达标准的患者与无结直肠癌家族史的患者之间,性别、结肠镜检查指征、免疫组化结果、病理特征或个人结直肠癌/其他林奇综合征相关肿瘤病史均无显著差异。Erell Guillerm等人对 16 例林奇样综合征患者进行肿瘤NGS分析,发现 6 例存在双体细胞突变,其中 1 例携带新发MSH2致病变异的嵌合体,该变异可传递给患者后代,对遗传咨询具有重要意义。Francesca Pirini等人通过综合遗传和表观遗传学方法探究林奇样综合征的致病机制,多基因panel分析发现,非错配修复(MMR)基因中的致病变异可能是林奇样综合征的易感因素;表观遗传学分析显示,存在靶向林奇综合征或DNA修复相关基因的表观变异,其中多数与范可尼贫血通路相关。
林奇样综合征的病因尚不明确,目前提出了四种致病机制:第一,MMR基因存在目前仍被归类为意义不明变异(VUS)的改变;第二,部分林奇样综合征患者可能实际是未确诊的林奇综合征病例,因为现有技术难以识别复杂或隐性突变。结构变异(如内含子区域突变、倒位、拷贝数变异[CNVs])在常规基因检测中很少被分析,但可能是这类患者的突变基础;第三,非MMR基因发生改变,如MUTYH、EXO1、POLE、POLD1、MCM、WRN、BARD1、RCF1、RPA1、MLH3、PPARG、CTC1、DCC、ALPK、PRKDC。已有研究在错配修复缺陷(dMMR)患者中发现MUTYH和POLE基因的胚系突变;第四,其他机制,如体质性表观遗传改变的存在,可导致错配修复缺陷表型。在无MMR基因改变但存在MMR蛋白表达缺失的散发性癌症中,其他分子机制(如体细胞致癌基因改变或MMR系统外的表观遗传事件)可能驱动dMMR/MSI表型,这类可能为散发性起源的肿瘤或可被排除在林奇样综合征分类之外,但由于缺乏区分这类病例的标准化流程,该分类方式仍存在争议。将患者归类为散发性或遗传性需要结合临床实践、全面的家系分析和长期随访,以评估结直肠癌/林奇综合征相关癌症的发病率差异。
本文报道了 4 例疑似林奇样综合征(LLS)病例,尽管样本量较小,但这些病例较为罕见,体现了林奇样综合征的临床异质性和机制复杂性。其中 2 例分别由肿瘤细胞内的错配修复(MMR)系统体细胞突变或POLE基因突变导致,无明确的遗传性疾病家族史;另外 2 例有肿瘤家族史,但未获取家属临床样本进行系统的胚系突变检测。病例3和4因检测到疑似致病变异,被诊断为非典型表型林奇综合征。这些发现表明,林奇相关综合征(包括林奇样综合征和非典型表型林奇综合征)的诊断高度依赖胚系突变检测结果,家属样本的缺失或检测到的变异性质差异会直接影响诊断分类的准确性。
参考文献:
Cheng B, Liu S, Ding S, Quan L, Liu J, Xu L, Zhao H, Guo J and Sun S (2025) Pathological diagnosis experience and literature review of four cases suspected Lynch-like syndrome. Front. Oncol. 15:1608253. doi: 10.3389/fonc.2025.1608253