深度解析医学证据,DeepEvidence为你支撑决策
Castleman病(CD)是一组至少包含 4 种淋巴增殖性疾病的复杂疾病谱,其中以单中心型CD最为常见。从形态学上,单中心型CD可进一步分为透明血管型和混合/浆细胞型变体。惰性T淋巴母细胞增殖(iT-LBP)是一种良性的胸腺外T淋巴母细胞扩增性病变,有时可与Castleman病相关。iT-LBP病例无形态学异型性或破坏性生长模式,不存在单克隆性或重现性遗传学异常证据,被认为是反应性病变。本文报告一例 49 岁女性患者,其发现盆腔肿块。穿刺活检显示为间质丰富的透明血管型单中心Castleman病。两年后肿块增大,切开活检可见不成熟淋巴母细胞弥漫性增殖,表达TdT、CD4和CD8,免疫表型无证据显示存在异常T细胞或B细胞群。形态学上无Castleman病的证据,但背景中可见梭形细胞增殖。二代测序检测到PDGFRB N666S突变,提示存在残余Castleman病。本文报告此病例是因为其凸显了Castleman病与iT-LBP之间的已知关联,而PDGFRB突变的检测支持iT-LBP很可能起源于透明血管型Castleman病的解读。
背 景
Castleman病(CD)以Benjamin Castleman的名字命名,他首次描述了CD的形态学特征,目前该病被公认为是一组至少包含 4 种淋巴增殖性疾病的疾病谱。Castleman病可分为单中心型和多中心型,其中单中心型CD最为常见。单中心型CD可表现出一系列形态学特征,也可进一步分为透明血管型和混合/浆细胞型变体。目前单中心型CD的发病机制在很大程度上尚不明确。已评估的罕见病例显示存在细胞遗传学异常,常涉及7号染色体,以及复杂核型。Chang及其同事使用人类雄激素受体检测法提出,透明血管型CD是一种起源于淋巴结基质细胞的良性肿瘤。也有研究报道了单中心型CD中的基因突变,其中PDGFRB突变最为常见,在高达 10% 的病例中存在。
惰性T淋巴母细胞增殖(iT-LBP)是一种罕见的临床良性淋巴增殖性疾病,其特征为前体T淋巴母细胞的多克隆增殖。2022 年,Saglam及其同事回顾了相关文献,总结了 45 例iT-LBP病例。后续也有相关病例报道,但我们估计迄今为止已报道的iT-LBP病例不超过 60 例。正如其他研究的定义,iT-LBP是TdT阳性、CD3阳性的中小体积T细胞的胸腺外扩增,这些细胞可形成密集的细胞簇或片层,生长模式无破坏性,无异常免疫表型或单克隆T细胞群的证据,与胸腺上皮无关,且临床行为呈惰性。iT-LBP不累及骨髓或纵隔。迄今为止,iT-LBP被认为是无重现性遗传学异常的反应性多克隆病变过程。
已有报道显示iT-LBP可与多种疾病合并发生。其中两种更常见的关联疾病包括单中心型CD和滤泡树突细胞肉瘤。iT-LBP也可与淋巴结T滤泡辅助细胞淋巴瘤,以及包括肝细胞癌和腺泡细胞癌在内的癌种相关。本文报告了一例表现为盆腔肿块的富间质型CD病例,该病变在两年的病程中进展为iT-LBP。在确诊iT-LBP时检测到了PDGFB突变。研究人员回顾了关于CD与iT-LBP相关性的文献,也讨论了PDGFRB突变的存在意义,以及iT-LBP的鉴别诊断。
病 例
患者女,49 岁,有皮肤基底细胞癌(已行切除术)、甲状腺乳头状癌(已行全甲状腺切除术及中央区颈清扫术,分期为T1aN1a,I期)病史,同时存在乳腺和卵巢良性病变,本次因偶然发现右侧盆腔肿块就诊评估。正电子发射断层显像/计算机断层扫描(PET/CT)显示,右盆腔后部存在一个 6.5×5.2 cm的强化病变,伴氟脱氧葡萄糖(FDG)中度摄取。粗针穿刺活检显示,取出的淋巴组织碎片中,滤泡约占标本的 10%。滤泡内淋巴细胞耗竭,可见少量透明血管病变。但病变大部分由丰富的间质成分构成,无异常滤泡树突细胞,也未见梭形细胞增殖(图1)。
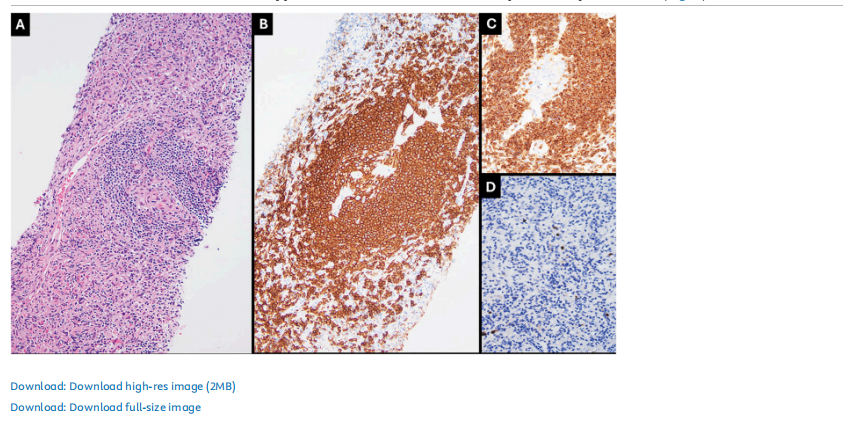

▲图1 Castleman病,透明血管型,富含间质
流式细胞术免疫表型分析未显示存在单型性B细胞或异常T细胞群的证据。免疫组化分析显示,滤泡富含CD20和PAX5阳性的B细胞,伴CD10和BCL6阳性、但BCL2阴性的小型生发中心。CD21染色可突显滤泡树突状细胞网。浆细胞的kappa和lambda轻链表达呈多型性。散在的背景T细胞CD3阳性。少量(<1%)散在的小淋巴细胞末端脱氧核苷酸转移酶(TdT)表达阳性。CD15、CD30、CD68、ALK、EMA和人类疱疹病毒8型(HHV8)检测结果均为阴性。Ki-67在 <5% 的细胞中呈阳性。EB病毒编码RNA(EBER)原位杂交分析为阴性。以上结果支持富间质亚型透明血管型CD的诊断,无恶性肿瘤证据。在后续两年中,PET/CT扫描显示盆腔肿块逐渐、持续增大,最终达到 8.5×6.3 cm。患者接受了剖腹探查术,并行次全子宫切除术、双侧输卵管卵巢切除术,以及盆腔侧壁肿块切开活检。由于肿块广泛累及血管,判定无法切除。对盆腔侧壁切开活检标本的组织学评估(图2)显示,中小体积淋巴样细胞弥漫性增殖,与大量梭形细胞混合存在于纤维化间质中。淋巴样细胞呈线性排列,可见极少量小型生发中心(占标本 <5%)。细胞学层面,淋巴样细胞核形圆、染色质细致、胞质稀少,无细胞异型性。免疫组化分析(图2)显示,淋巴样细胞呈现不成熟T细胞免疫表型,TdT(强阳性)、CD1a、CD2、CD3、CD4、CD5、CD7、CD8和BCL2均为阳性,CD10表达不一,CD31呈弱而局灶性表达。淋巴样细胞LMO2、CD20、CD21、CD23、CD30、CD34、CD35、CD68、CD79a、CD138、CD163、PAX5、kappa、lambda、细胞周期蛋白D1、ALK、细胞角蛋白(AE1/AE3)、平滑肌肌动蛋白(SMA)、结蛋白、肌细胞生成素、MYOD1、EMA、S100蛋白、SOX10、聚集素、CXCL13和IgG4均为阴性。混杂的梭形细胞SMA、CD163(部分)和聚集素(弱阳性)表达阳性。血管CD31和CD34阳性。
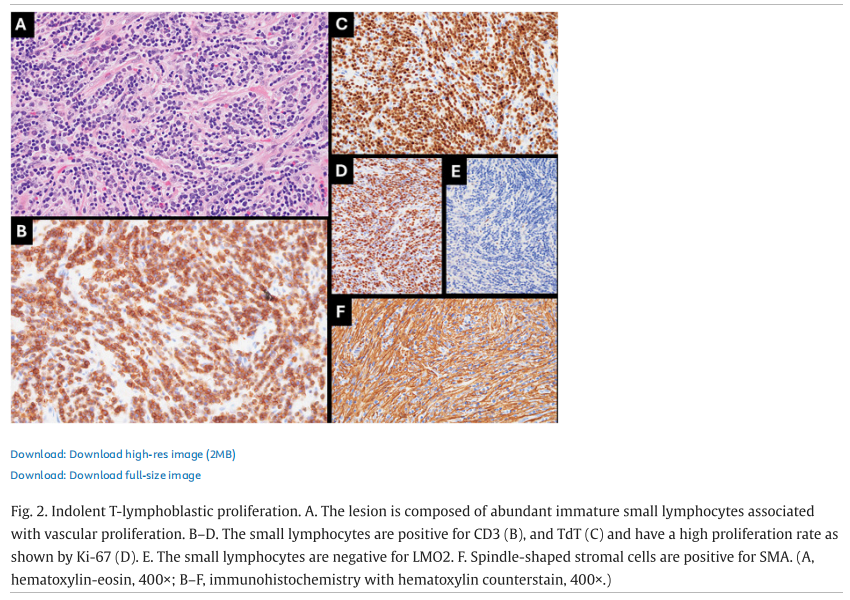
▲图2 惰性T淋巴母细胞增殖
对该标本进行了TRG分析以评估克隆性,结果呈多克隆模式,未检测到克隆性TRG重排。使用TSO 500检测试剂盒进行二代测序分析,结果显示存在PDGFRB N666S突变,变异丰度为 6.7%。总体,以上结果支持惰性T淋巴母细胞增殖的诊断。形态学上无Castleman病、淋巴瘤或肉瘤的证据。
讨 论
单中心型CD病例大多表现出透明血管(HV)亚型的形态学特征。该亚型的特点是淋巴滤泡和滤泡间间质出现特征性改变。尽管所有HV-CD病例均存在这些改变,但不同病例的滤泡和间质成分占比存在差异,部分病例以滤泡改变为主,另一部分则以间质改变为主。HV-CD的滤泡改变包括体积较大的滤泡,可包含 2 个或更多生发中心(即所谓的“孪生滤泡”);套区增宽,由小淋巴细胞呈层状线性排列(即所谓的“洋葱皮”样改变);以及淋巴细胞耗竭的生发中心,被硬化的放射状小动脉穿透(即所谓的透明血管或“棒棒糖”样病变)。Danon等人将间质占比 >50% 定义为间质为主型,这类病例被命名为富间质亚型HV-CD。该亚型更常发生于盆腔,与本文报告的患者发病部位一致。这类病例的间质由大量小淋巴细胞、增多的血管以及梭形细胞增殖构成,梭形细胞通常表达SMA,部分病例可表达滤泡树突细胞分化标志物。富间质亚型HV-CD的识别难度较高,尤其是在小型穿刺活检标本中。
目前已知CD与iT-LBP相关。在已报道的iT-LBP病例中,有 21 例记录存在CD或CD样改变。其中大多数病例的CD成分为单中心型透明血管亚型,但至少有 2 例多中心型CD合并iT-LBP的报道。大多数单中心型HV-CD病例存在易于识别的HV-CD典型滤泡改变,但目前已报道 2 例合并iT-LBP的富间质型HV-CD,包括本病例。
文献报道的 21 例CD相关iT-LBP病例的人口统计学数据总结于表1。患者发病中位年龄为37岁(范围 13-71 岁),包括 11 例女性、9 例男性,1 例未报告性别。淋巴结是最常受累的解剖部位,共 12 例患者受累,包括颈部(n = 4)、腹股沟(n = 3)、腹膜后(n = 1)、主动脉旁(n = 1)、腋窝+腹股沟(n = 1)、全身淋巴结肿大(n = 1)以及未明确部位(n = 1)。其他受累部位包括盆腔(n = 4)、腹膜后(n = 3)和肾上腺(n = 1)。19 例合并透明血管型CD,1 例合并特发性多中心型CD,1 例合并HHV8阳性多中心型CD。除本病例外,此前报道的所有病例均未进行突变分析。
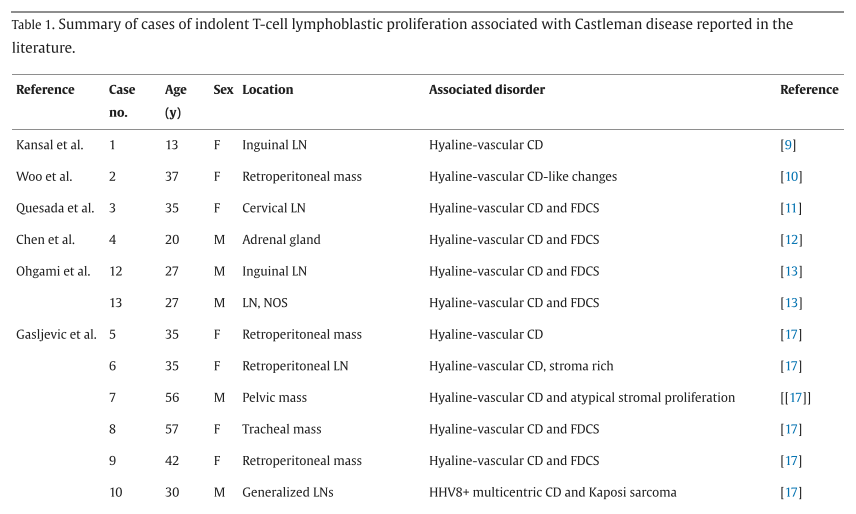
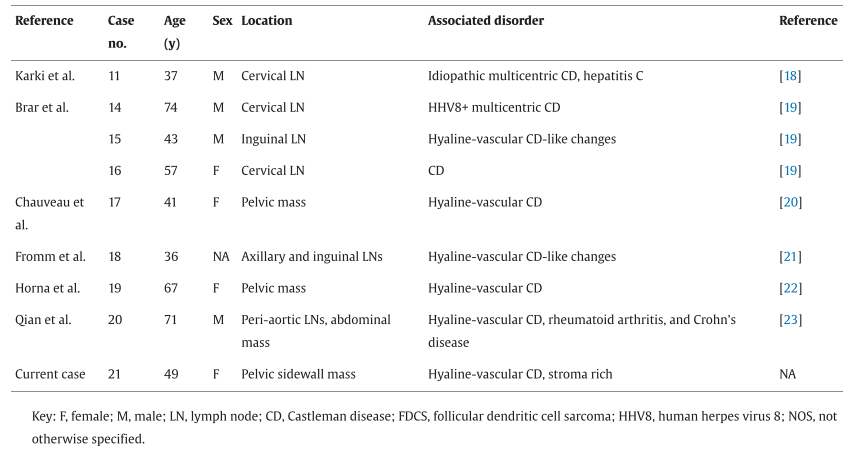
▲表1 文献报道的与Castleman病相关的惰性T细胞淋巴母细胞增殖病例总结
已有研究在少数单中心型CD病例中报道了PDGFRB突变,报道的突变频率为 10%-17%,迄今为止最常见的突变类型为PDGFRB p.N666S。在本研究报告的病例中,该突变在iT-LBP病变中被检出,且病变背景中存在梭形细胞。目前普遍认为iT-LBP是一种非克隆性增殖病变,这一结论主要基于T细胞受体基因重排研究显示其为多克隆T细胞群,且此前尚无iT-LBP病例存在基因突变的报道。本病例中PDGFRB突变的检出提示,活检标本中的梭形细胞代表残余的富间质型HV-CD,且正是这些梭形细胞携带PDGFRB突变,活检标本中该突变的低变异等位基因频率也支持这一解读。
血小板衍生生长因子受体β(PDGFRB)是一种位于5号染色体长臂32区的受体酪氨酸激酶,可通过RAS–MAPK、PI3K–AKT和JAK–STAT通路向下游传递信号,调控间充质/血管周细胞(周细胞、平滑肌细胞)的生物学功能及血管生成。其自身抑制作用由近膜区和激活环介导,激活性改变会破坏这种调控,产生不依赖配体的信号传导。在癌组织中,PDGFRB更多表达于间质周细胞和成纤维细胞,而非肿瘤细胞本身,在肿瘤组织中可促进血管生成并参与形成免疫抑制微环境。一般认为PDGFRB突变会导致PDGFRB持续激活,目前PDGFRB激活如何参与HV-CD的发病机制尚不明确。
iT-LBP的病变细胞具有特征性的免疫表型,与正常皮质胸腺细胞高度相似。病变细胞通常表达TdT、胞质CD3以及CD2、CD5、CD7等泛T细胞抗原,CD4和CD8共表达较为常见,不过也有极少数CD4/CD8双阴性病例的报道。CD1a常呈阳性,进一步支持这些淋巴母细胞具有皮质胸腺细胞的表型特征。本病例的免疫表型结果与上述模式一致,但存在一个不典型特征:本病例中的淋巴母细胞BCL2阳性。在正常胸腺皮质中,BCL2通常为阴性,而髓质胸腺细胞BCL2为阳性,这一发现提示iT-LBP中的T细胞并非严格意义上的皮质胸腺细胞,或未遵循正常T细胞成熟过程。iT-LBP的主要鉴别诊断为T细胞急性淋巴细胞白血病(ALL)或淋巴母细胞淋巴瘤(LBL)。
iT-LBP和T-ALL/LBL的细胞免疫表型极为相似,且Ki-67评估的增殖率在两类病变中均较高。LMO2是有助于该鉴别诊断的一个免疫表型标志物:Brar及其同事在小样本研究中报道,13 例T-LBL病例中有12例LMO2阳性,而评估的7例iT-LBP病例全部为LMO2阴性,本研究讨论的病例同样LMO2阴性。此外,与大多数T-ALL/LBL病例不同,本病例无克隆性TRG重排。其他支持T-ALL/LBL、但不存在于iT-LBP的特征还包括:淋巴母细胞存在明显核折叠和/或异型性、异常T细胞免疫表型、骨髓和/或纵隔受累,以及侵袭性临床行为。
综上,本文报告了一例极为罕见的单中心型Castleman病进展为惰性T淋巴母细胞增殖的病例,后者病变携带PDGFRB p.N666S突变。本病例印证了单中心型透明血管型CD与iT-LBP之间的已知关联,进一步支持部分透明血管型CD病例存在PDGFRB突变的结论。同时本病例也凸显了富间质型HV-CD好发于盆腔的特点,以及诊断富间质型HV-CD的难度,尤其是在小型穿刺活检标本中。
参考文献:
Al-Rusan O, Ward DE, Chang CC, Shen Q, Medeiros LJ. From the archives of MD Anderson Cancer Center: Stroma-rich hyaline vascular Castleman disease followed by indolent T-lymphoblastic proliferation and detection of PDGFRB mutation. Ann Diagn Pathol. 2026 Feb;80:152573. doi: 10.1016/j.anndiagpath.2025.152573. Epub 2025 Oct 5. PMID: 41066910.