深度解析医学证据,DeepEvidence为你支撑决策
脓毒症作为一种死亡率高、威胁全球的健康问题,其并发的脓毒症相关性脑病(SAE)会进一步增加患者的死亡风险。近期研究表明,脓毒症后肠道免疫细胞可迁移至肺部,但其对中枢神经系统的影响尚不明确。本研究发现,脓毒症可诱导IL-7Rhigh CD8low γδ T17细胞从小肠迁移至脑膜,并在脑膜中分泌IL-17A,进而损害雄性小鼠小胶质细胞的线粒体功能,激活cGAS-STING-C1q通路。该过程中,STING蛋白K150位点的泛素化受到抑制,导致STING积聚,并使C1q标记的海马突触数量增加,这些突触随后被活化的小胶质细胞所修剪。4-辛基衣康酸(4-OI)通过抑制γδ T17细胞迁移并促进STING泛素化,减轻了过度的突触修剪,从而缓解SAE。本研究揭示了一个由小胶质细胞通过cGAS-STING-C1q通路介导突触修剪的潜在机制,阐明了肠源性γδ T17细胞向脑膜迁移在SAE中的关键作用,并强调了STING泛素化在调控C1q介导的过度突触修剪中的重要性。该研究成果于2025年7月发表在《Nature Communications》上。

研究者首先通过实验发现,脓毒症可诱导小肠中的IL-7Rhigh CD8low γδ T17细胞直接迁移至脑膜。为进一步验证其作用,作者在γδ T细胞特异性敲除IL-7R的小鼠模型中发现,阻断IL-7R信号不仅显著减少了脑膜中致病的γδ T17细胞数量,还能够逆转脓毒症引起的树突棘丢失与排列紊乱、突触超微结构损伤及线粒体功能障碍。同时,小鼠在Y迷宫、水迷宫和新物体识别等行为学测试中的认知与记忆缺陷也得到明显改善。以上结果表明,IL-7Rhigh CD8low γδ T17细胞可加重SAE中的认知功能障碍。

图1.小肠IL-7Rhigh CD8low γδ T17细胞向脑膜迁移加剧SAE
明确小肠来源的IL-7Rhigh CD8low γδ T17细胞向脑膜迁移可加重SAE后,研究者进一步探讨了其潜在的分子机制。结果显示,脑膜中超过50%的IL-17A由γδ T17细胞产生,该细胞因子可诱导小胶质细胞线粒体膜电位下降及线粒体肿胀,进而激活cGAS-STING信号通路。体外共培养实验证实,敲低γδ T细胞中的IL-17A表达可减轻原代小胶质细胞的线粒体损伤,并降低cGAS、STING及补体C1q的水平。在体内实验中,海马区过表达IL-17A可上调cGAS和STING的表达,增加C1q水平,同时下调突触蛋白PSD95和SYN的表达,并增强小胶质细胞对突触的吞噬作用,最终导致认知功能损害;而中和IL-17A或条件性敲除IL-17R则可逆转上述改变。进一步采用STING抑制剂H151、直接中和C1q或使用PLX3397清除小胶质细胞,均能显著减少突触修剪,改善脓毒症小鼠的行为学缺陷。综上所述,γδ T细胞来源的IL-17A通过损伤小胶质细胞线粒体、激活cGAS-STING通路并促进C1q介导的突触吞噬,构成脓毒症相关认知障碍的核心机制,该通路中的关键分子可能成为潜在的治疗靶点。
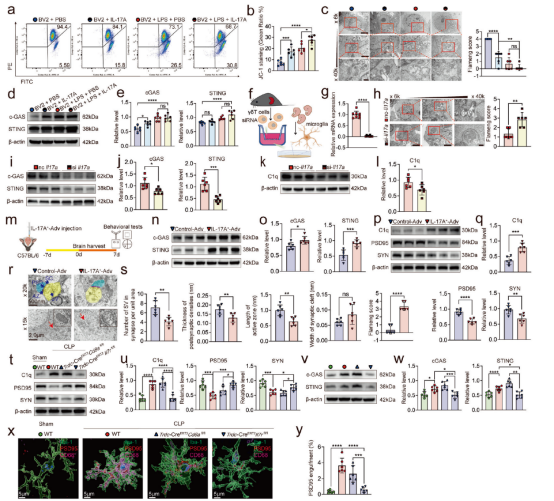
图2. γδ T 细胞产生的 IL-17A 会损伤小胶质细胞线粒体并激活 cGAS-STING-C1q 通路。
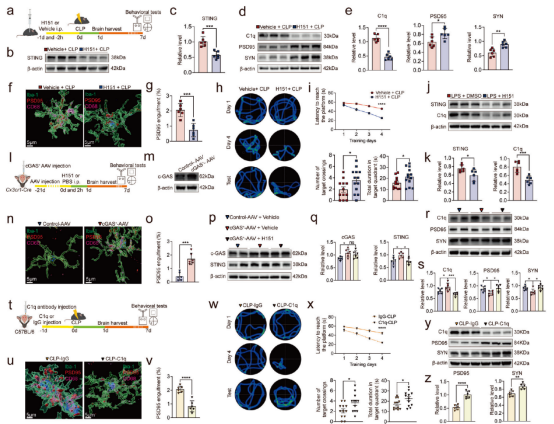
图3. STING 增加会促进 C1q 依赖性突触修剪。
本研究进一步探讨了SAE中STING蛋白的调控机制,发现泛素-蛋白酶体系统可通过K150位点的泛素化修饰调控STING的降解。在脓毒症条件下,小胶质细胞中STING的K150泛素化水平显著降低,导致STING蛋白积聚。进而增强小胶质细胞对PSD95的吞噬能力,同时增加C1q表达并降低突触蛋白PSD95和SYN的水平,诱导线粒体超微结构损伤和突触修剪,最终导致小鼠出现严重的学习与记忆缺陷。机制研究表明,E3泛素连接酶RNF5与STING直接相互作用,并介导其K150位点的泛素化;而在脓毒症条件下,二者的相互作用减弱。进一步实验显示,敲低RNF5可降低STING泛素化水平,而过表达RNF5则促进STING泛素化、降低C1q表达、恢复突触蛋白水平并改善认知功能。综上所述,小胶质细胞中RNF5介导的STING K150泛素化是防止过度突触修剪及认知功能障碍的关键调控机制。
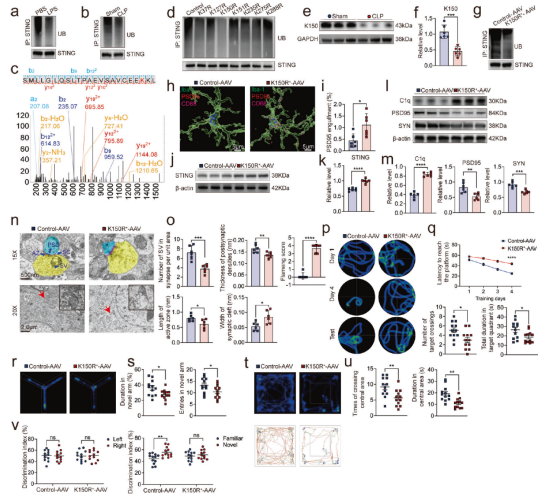
图4. K150 位点对于小胶质细胞中STING的泛素化至关重要。
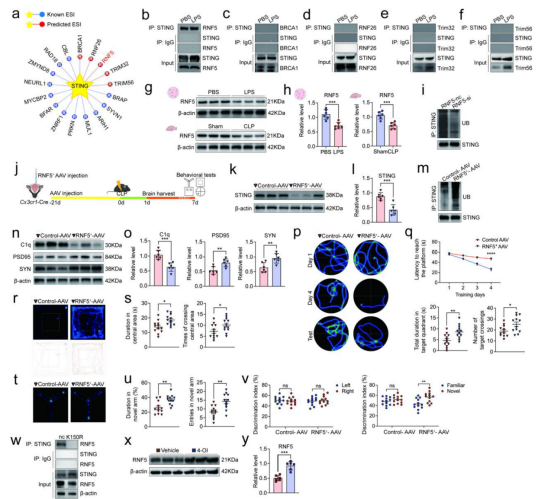
补充图 7. RNF5 增强STING泛素化减轻SAE。
进一步的蛋白质组学分析发现,脓毒症后小肠和海马中衣康酸合成酶ACOD1显著上调,且小胶质细胞是脑内衣康酸的主要来源。采用可透过血脑屏障的衣康酸衍生物4-辛基衣康酸(4-OI)治疗能够增强肠道屏障完整性,显著抑制小肠γδ T17细胞向脑膜的迁移,减少IL-17A的释放,并降低cGAS-STING信号通路及补体C1q的水平,从而减轻海马区的突触损伤、线粒体功能障碍以及小胶质细胞对突触的异常吞噬。分子对接及突变实验表明,4-OI主要通过结合cGAS蛋白的Ser366位点发挥作用。在功能层面,4-OI可恢复CA1区锥体神经元的突触传递效能,改善脓毒症小鼠的脑血流量和葡萄糖代谢,并在Y迷宫、水迷宫及新物体识别等行为学测试中显著逆转认知缺陷。此外,4-OI还能增强肠道屏障完整性、降低全身炎症反应并提高生存率,且在CLP和CASP两种脓毒症模型中均表现出稳定的神经保护效果。综上所述,4-OI通过靶向cGAS-STING-C1q轴并阻断“肠道-脑膜”免疫细胞迁移通路,是一种具有前景的SAE治疗候选药物。
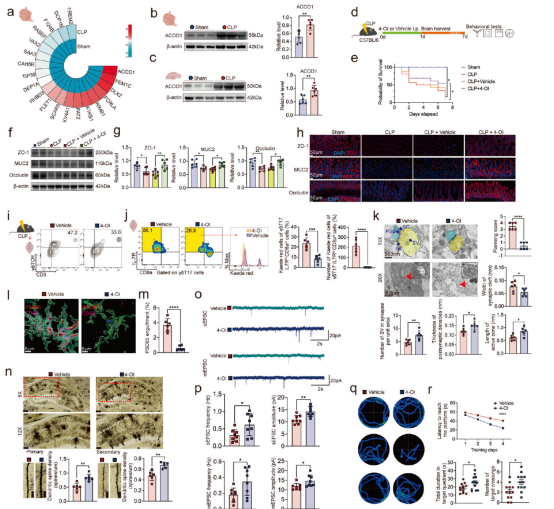
图5.4-OI缓解SAE
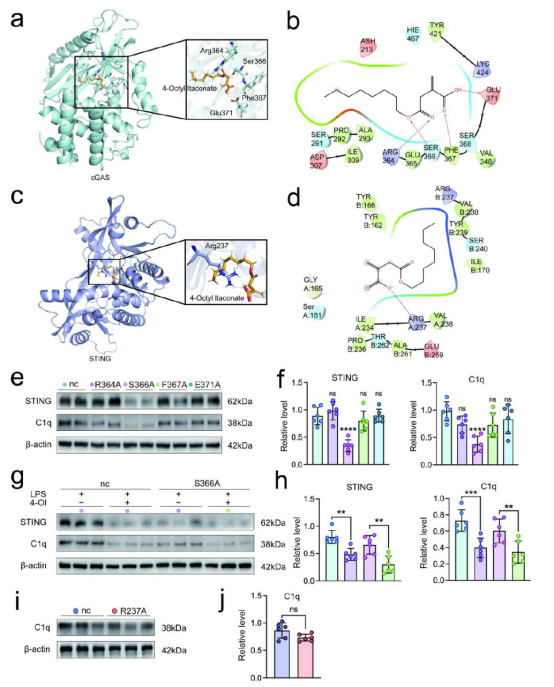
补充图14. cGAS与4-OI的相互作用和结合位点。
使用ACOD1基因敲除小鼠的研究发现,该类小鼠在CLP术后24小时内无法存活,且在术后16小时即表现出突触与线粒体损伤加重、小胶质细胞突触吞噬增强、STING与C1q表达升高以及突触蛋白减少等病理改变,而4-OI治疗可缓解上述异常。进一步利用AAV在Cx3cr1-Cre小鼠的小胶质细胞中特异性沉默ACOD1,同样导致cGAS-STING通路激活、K150位点介导的STING泛素化减少、C1q水平升高、突触蛋白PSD95和SYN表达下降、小胶质细胞对突触的吞噬增强,以及认知功能障碍加重。综上所述,小胶质细胞中的ACOD1通过促进K150位点介导的STING泛素化,抑制cGAS-STING-C1q通路,从而防止过度的突触修剪;其缺失或抑制则会破坏这一保护机制,进而加剧脓毒症相关的突触损伤和认知障碍。
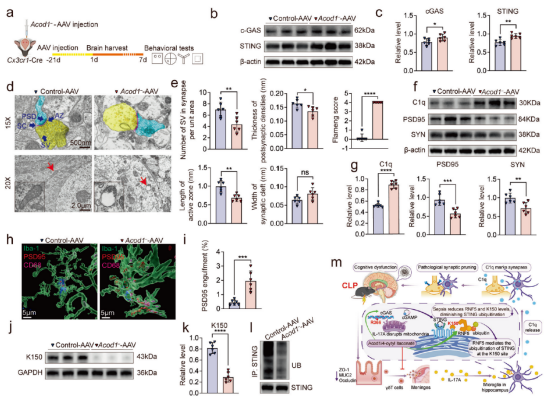
图6. 小胶质细胞中ACOD1的敲除加重认知功能障碍
醉翁之艺点评
本研究首次揭示了小肠来源的IL-7Rhigh CD8low γδ T17细胞在脓毒症相关脑病(SAE)中的关键致病机制。研究人员发现,脓毒症可诱导这群γδ T17细胞从小肠迁移至脑膜,并通过分泌IL-17A损伤小胶质细胞的线粒体,进而激活cGAS-STING信号通路。脓毒症状态下STING蛋白K150位点的泛素化受到显著抑制,导致STING异常积聚,从而促进C1q的释放。C1q作为“吃掉我”信号标记海马区的突触,使其最终被活化的小胶质细胞过度修剪,引发突触丢失与认知功能障碍。该研究首次将肠道免疫细胞迁移、线粒体损伤、STING信号调控与补体介导的突触修剪串联成一条完整的“肠-脑轴”致病链条,为SAE的发病机制提供了全新视角。
在治疗探索方面,研究者验证了4-辛基衣康酸(4-OI)的双重保护作用:一方面抑制γδ T17细胞向脑膜的迁移,另一方面促进STING K150位点的泛素化降解,从而阻断cGAS-STING-C1q通路的过度激活,显著减轻突触修剪并改善脓毒症小鼠的认知行为。作为一种可透过血脑屏障的衣康酸衍生物,4-OI展现出良好的神经保护与抗炎效果。尽管该研究目前仅在雄性小鼠中验证,且临床相关性尚需进一步确认,但其清晰的机制解析和明确的干预靶点,为SAE乃至阿尔茨海默病、多发性硬化等神经炎症疾病的免疫治疗开辟了新的方向。