首页 > 医疗资讯/ 正文
2025年6月,中华人民共和国国家卫生健康委员会发布了《第二批罕见病目录》中86个病种的诊疗指南,旨在进一步提高我国罕见病诊疗规范化水平,本文特摘录其中关于阿拉杰里综合征(ALGS)的相关内容,供临床参考。
1概述
ALGS是一种常染色体显性多系统受累的遗传病,由Daniel Alagille等于1969年首次报道。该病估计的发病率为1/50 000~1/30 000,无明显性别差异,主要临床特征有慢性胆汁淤积、心脏杂音、蝴蝶椎骨、角膜后胚胎环、特殊面容和肾脏畸形,是合并肝外器官受累的儿童慢性胆汁淤积的最常见原因之一。
2病因和流行病学
ALGS由高度保守的NOTCH信号通路缺陷导致。NOTCH信号通路调控细胞组织发育、稳态和修复。已知NOTCH信号通路存在5种配体(JAG1、JAG2、DLL1、DLL3和DLL4)和4种受体(NOTCH1、NOTCp、NOTCp和NOTCp)。文献报道94%~96%的ALGS由JAG1基因突变所致,2%~3%由NOTCp基因突变引起,尚有2%~4%患者未找到致病突变基因(推测由于检测技术的限制所致)。JAG1和NOTCp在人体发育的各个阶段及多种组织中均有表达。在人类胚胎的肺动脉、主动脉、心脏远端流出道、后肾、胰腺、神经管、视神经泡及耳泡均可检测到JAG1表达。JAG1表达广泛,其功能缺陷可相应导致多器官出现临床表型。JAG1-NOTCp信号轴对于正常胆管的形成十分必要,JAG1或NOTCp基因变异引起的该信号轴功能缺陷可导致胆管异常,所以ALGS患者肝组织活检病理的典型表现是小叶间胆管减少或缺失。
最初估算的全球ALGS的发病率在1/70 000。随着分子诊断的普及,估算的ALGS发病率提高到1/50 000~1/30 000。国内缺乏相关的流行病学数据,截至2021年10月,复旦大学附属儿科医院共诊断223例ALGS病例,是伴有多系统受累的儿童胆汁淤积症最常见遗传性病因。
3临床表现
ALGS的显性率约 98%,但个体的表现度可有很大差别,因此表型有高度变异性。该病可累及多个器官,临床以肝脏、心脏、骨骼、眼睛异常及特殊面容表现最常见。截至目前,多项临床研究显示基因型与表型之间无显著关联。
3.1 肝脏表现
常表现为不同程度的胆汁淤积,导致慢性胆汁淤积性肝病。黄疸是该病最主要的表现之一,多数在婴儿早期,尤其在新生儿期即可出现高结合胆红素血症,呈阻塞性黄疸表现。大约50%的患者黄疸持续整个婴儿期,部分患儿黄疸可随年龄增长逐渐缓解。瘙痒是ALGS的突出表现。ALGS患者可有严重的高脂血症,以血清中胆固醇升高最明显。严重者可见多发性黄瘤,通常在出生后数年内逐渐增多,随胆汁淤积改善可消失。凝血功能障碍常见,多数注射维生素K1可纠正。肝脏表型的严重程度是影响ALGS患者预后的主要原因。
3.2 心脏表现
心脏杂音是ALGS患者的常见体征,杂音主要因肺动脉流出道或外周肺动脉的狭窄引起。肺动脉狭窄可单独存在,也可合并法洛四联症、室间隔缺损、房间隔缺损等。
3.3 骨骼表现
ALGS患者可有脊椎异常,主要表现为蝶形椎骨,多见于胸椎。蝶形椎骨通常不表现出临床不适症状。33%~87%的患者可出现特征性的蝶形椎骨。其他骨骼异常包括尺桡骨融合、脊柱片显示椎体中央透亮等。此外,ALGS综合征患者可因缺乏维生素D发生代谢性骨病、骨质疏松症及病理性骨折。
3.4 眼部表现
角膜后胚胎环是凸出中心位的Schwalbe环,常出现在角膜内皮和色素层小梁组织的交界处,是最具有特征性的眼部表现。角膜后胚胎环可见于56%~95%的患者,但是8%~15%的正常人也可出现,所以角膜后胚胎环单独出现诊断价值有限。其他眼部异常包括青光眼、角膜巩膜发育不全、中胚层发育不全、视神经乳头异常等。该病患者的眼部异常很少出现临床症状。
3.5 面部表现
ALGS患者的面部特征为前额突出、眼球深陷伴眼距中度增宽、尖下颌、鞍形鼻并前端肥大等。小婴儿以前额突出多见,随年龄增长,其他各项特征逐渐突出。成年人前额突出不太明显,但下颌突出更明显。其他面部特征还有耳朵大等。
3.6 肾脏表现
ALGS患者可有孤立肾、小型肾、多囊肾等肾发育异常,部分患儿可出现肾小管酸中毒。
3.7 其他表现
ALGS患者可出现血管发育异常,可表现为烟雾病、肾动脉、腹腔动脉、肠系膜上动脉、锁骨下动脉等异常。有研究证实超过30%的ALGS患者头颅MRI提示血管异常,这些患者大多无任何症状。其他的异常有声音尖、音调单一等。
许多患者的父亲或母亲可表现出ALGS的1项或1项以上临床特征,其中以角膜后胚胎环和心脏杂音最为常见,也有表现为婴儿期短暂的胆汁淤积、蝴蝶椎骨等。
4辅助检查
4.1 常规实验室检查
多数ALGS患者肝功能化验中可见血清胆红素、胆汁酸升高,GGT明显升高,血中转氨酶水平也不同程度升高,但肝脏合成功能常不受影响。部分患者肝脏表型轻,血清胆红素可在正常范围内,以转氨酶升高为主。凝血功能障碍常见,但多在注射维生素K1后可纠正。该病患者多伴有严重的高脂血症,尤其以血中胆固醇升高最明显。部分患者尿常规及血气分析可见肾小管酸中毒表现。
4.2 影像学检查
部分腹部超声可见肾脏发育不良、孤立肾等异常。心脏超声可见肺动脉狭窄等畸形。部分患儿行头颅MRI血管成像可见动静脉畸形。
4.3 肝组织病理
肝脏活检病理发现小叶间胆管减少或缺乏曾被认为是ALGS必不可少的特征。近年研究发现,部分患儿在婴儿早期可无小叶间胆管消失或减少,其小叶间胆管消失是在生后逐渐发生的。6月龄前进行肝脏穿刺活检,仅有约60%的患儿有小叶间胆管缺乏;6月龄后进行肝活检,95%的患儿可表现为小叶间胆管缺乏。部分病例可表现为汇管区的减少,部分病例汇管区可有炎症细胞浸润,早期纤维化常不明显。少部分病例在疾病早期可有小胆管的增生,此时与胆道闭锁鉴别非常困难。
5诊断
临床诊断的确立依赖于综合判断。经典的诊断标准为肝组织活检有肝内小叶间胆管数量减少或缺如,并具有至少包括慢性胆汁淤积、心脏杂音、蝴蝶椎骨、角膜后胚胎环和特殊面容等5个主要临床表现中的3个,并排除其他可能原因。现在肾脏异常也列为主要异常之一。如果肝活检不表现为肝内小叶间胆管数量减少或缺如,或由于某些成年轻症患者并未进行肝活检,修订的诊断标准认为符合4个或以上主要标准也可诊断。如果已知有JAG1/NOTCp基因突变或阳性家族史时,2个主要标准通常即可确诊。
6鉴别诊断
ALGS患者血清GGT升高明显,因此需要和伴有GGT 升高的各种婴儿期胆汁淤积症相鉴别。由于ALGS患者脊椎、眼睛和肾脏异常多无显著的临床表现,特征性面容在婴儿早期也不显著等原因,ALGS与其他原因引起的胆汁淤积症鉴别也有一定难度。ALGS早期诊断面临的最大挑战是如何与胆道闭锁相鉴别。由于胆道闭锁需要尽早手术治疗,而有报道若把ALGS误诊而进行葛西手术可使预后变差,因此及时进行鉴别诊断尤为重要。婴儿高GGT胆汁淤积症患儿行脊柱摄片、心脏超声及眼科检查有助于鉴别ALGS与胆道闭锁。此外,肝穿刺组织活检对鉴别诊断也有很大帮助。胆道闭锁的特征是小胆管显著增生,而ALGS虽然在早期可不存在肝内胆管消失或减少,但也少见显著小胆管增生。其他需要鉴别的以多系统受累为特征的疾病包括威廉姆斯综合征、歌舞伎(Kabuki)综合征等。
7治疗
目前尚无特异的ALGS治疗手段,主要是支持治疗和对症处理。由于该病有多系统受累,因此良好的管理需要多学科专科医师参与。肝病方面主要面临的是胆汁淤积及其并发症,包括脂溶性维生素缺乏。通常需要常规补充脂溶性维生素,并通过维生素浓度或凝血酶原时间进行检测。利胆药可选用熊去氧胆酸,每天20~30 mg/kg,分次口服。最新批准的氯马昔巴特可用于治疗1岁及以上ALGS患者的胆汁淤积性瘙痒,推荐剂量为380 µg/kg,每日1次,早餐前30 min口服。应避免和胆汁酸结合树脂同时使用,或前后至少间隔4小时。建议使用前详细阅读说明书。
ALGS患者需要定期监测肝功能变化,必要时行肝移植。肝移植是ALGS终末期肝病患儿的有效治疗措施。
8诊疗流程(图1)
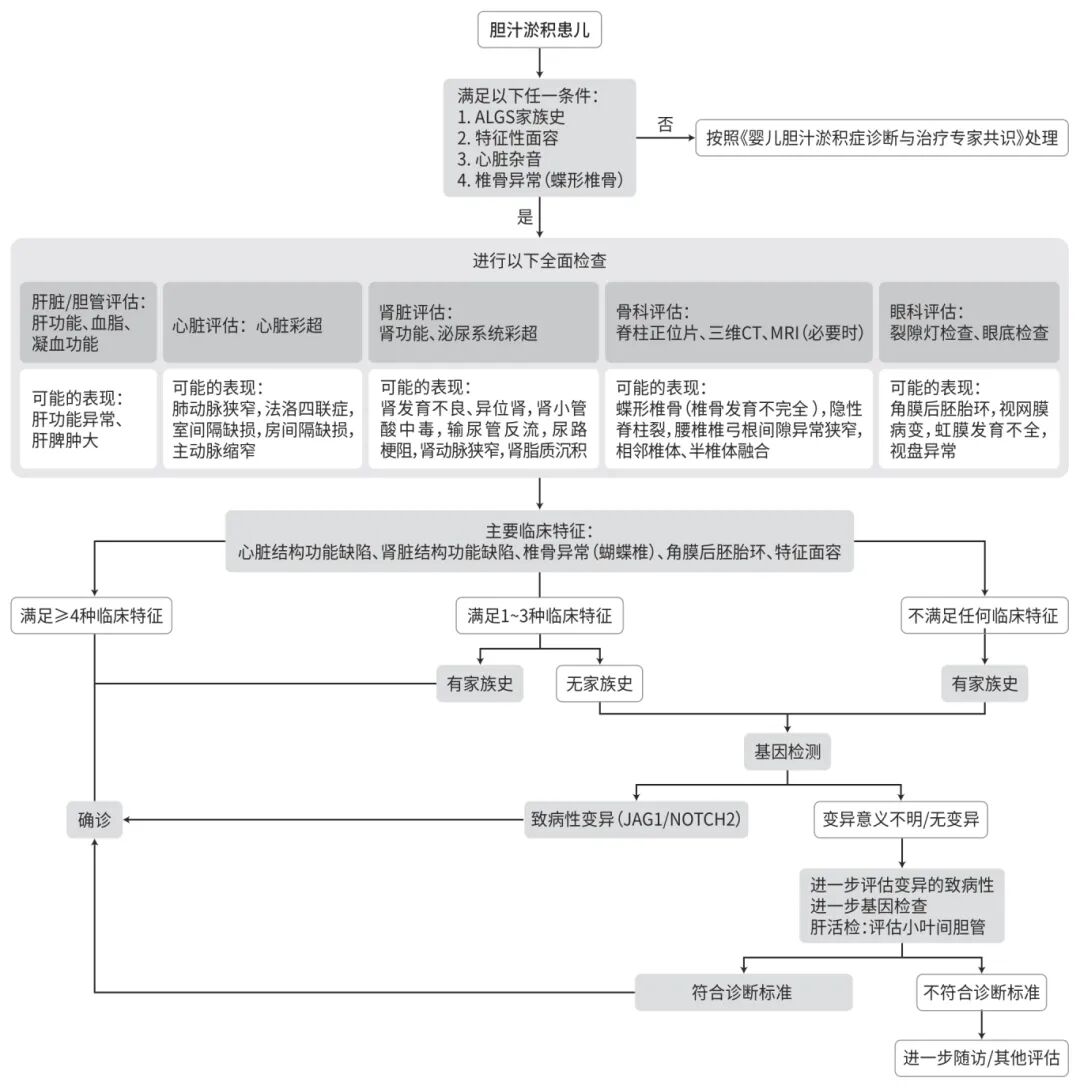
注: JAG1,Jagged1基因;NOTCp,Notch受体2基因。
图1 ALGS诊疗流程
全文下载
https://www.lcgdbzz.org/cn/article/doi/10.12449/JCp50907

- 搜索
-
- 1000℃Nutrients:真实世界数据,纤维肌痛患者的饮食与运动自适应规律
- 1000℃D-二聚体升高诊治与管理专家共识(2026)
- 1000℃专家论坛|文良志:门静脉血栓的诊断和治疗
- 1000℃首例儿童NF2驱动型胸膜间皮瘤,多方法学检测锁定NF2双等位基因失活和14/22号染色体缺失,提示与成人胸膜间皮瘤不同
- 1000℃打破误区:干扰素追求CHB功能性治愈,HBsAg为何“不降反增”?
- 1000℃迷惑性极强的肝内病灶!影像表现疑点重重,最终病理竟查出两种不同肝脏恶性肿瘤
- 1000℃指南共识|原发性肝癌分子靶向药物相关蛋白尿中西医结合诊疗专家共识
- 1000℃Diabetologia:意大利北部社区 1~100 岁人群胰岛自身抗体与乳糜泻 TGA-IgA 的年龄分布及检测方法学验证
- 精J Child Psychol Psychiatry:12种罕见神经发育障碍儿童沟通能力谱系
- 精研究发现:爱吃辣的人,心血管病和癌症死亡风险都会显著降低
- 精Nursing in Critical Care:别再指责护士了!ICU 里被遗漏的护理,根源在系统而非个人
- 精Acta Obstet Gynecol Scand:罕见病女性的妊娠并发症与母婴结局,一项单中心434种罕见病的回顾性队列研究
- 精【爱儿小醉】儿科患者术前对流层臭氧暴露与围手术期呼吸系统不良事件之间的关系:一项单中心回顾性队列研究
- 精eBioMedicine:牙龈下微生物组与脑健康存在连续关联梯度,牙周炎或成认知衰退可干预靶点
- 精军事医学研究院《自然·通讯》:自适应IrPtCu纳米酶水凝胶实现耐药菌感染伤口序贯治疗
- 精能够逆转萎缩性胃炎的两个中成药,该怎么选择?
- 荐同时性多发性原发性肺癌,左右病灶分别为EGFR和ALK阳性,考虑淋巴结肿大仅局限左肺门及血浆EGFR阳性,采用奥希替尼联合化疗
- 荐40岁女性同时罹患卵巢支持细胞-间质细胞瘤和透明细胞乳头状肾肿瘤,WES等基因检测竟为阴性
- 荐椎管内麻醉使用止血药突发气道痉挛的抢救流程解析
- 荐女子肝区无任何不适,影像提示复杂囊性病变,层层鉴别后锁定罕见胆管源性囊性肿瘤
- 荐“绘”真报告 | 病理考虑为中枢神经细胞瘤,检出脑室外神经细胞瘤的特征性变异FGFR1-TACC1融合,辅助鉴别诊断
- 荐8岁女童出现男性化症状,竟是形似「性索-间质肿瘤」的卵巢「无性细胞瘤」所致,少见KRAS/CDK4共扩增或与侵袭性有关
- 荐17例病例分析揭示常见于中年人的色素性室管膜瘤临床特征与预后,分子检测可助力临床精准诊疗
- 荐Lancet Oncol:结直肠癌腹膜转移,围手术期化疗并非必选项
- 标签列表
-
- 星座 (702)
- 孩子 (526)
- 恋爱 (505)
- 婴儿车 (390)
- 宝宝 (328)
- 狮子座 (313)
- 金牛座 (313)
- 摩羯座 (302)
- 白羊座 (301)
- 天蝎座 (294)
- 巨蟹座 (289)
- 双子座 (289)
- 处女座 (285)
- 天秤座 (276)
- 双鱼座 (268)
- 婴儿 (265)
- 水瓶座 (260)
- 射手座 (239)
- 不完美妈妈 (173)
- 跳槽那些事儿 (168)
- baby (140)
- 女婴 (132)
- 生肖 (129)
- 女儿 (129)
- 民警 (127)
- 狮子 (105)
- NBA (101)
- 家长 (97)
- 怀孕 (95)
- 儿童 (93)
- 交警 (89)
- 孕妇 (77)
- 儿子 (75)
- Angelababy (74)
- 父母 (74)
- 幼儿园 (73)
- 医院 (69)
- 童车 (66)
- 女子 (60)
- 郑州 (58)