首页 > 医疗资讯/ 正文
摘要
遗传性骨骼肌通道病是一组因骨骼肌离子通道基因突变引发的异质性遗传性疾病。根据临床表现,遗传性骨骼肌通道病被分为两大类:非营养不良性肌强直和周期性麻痹。非营养不良性肌强直包括先天性肌强直、先天性副肌强直和钠通道肌强直等。周期性麻痹分为低钾型、正常血钾型、高钾型和Andersen-Tawil综合征。非营养不良性肌强直和周期性麻痹在临床表型和分子层面上存在重叠,少数患者同时表现出非肌营养不良性肌强直和周期性麻痹表型,这进一步表明遗传性骨骼肌通道病在临床谱系中是一个连续的过程。本研究对骨骼肌离子通道病的分类、临床表现、诊断标准、基因病理分型、致病机制、治疗方法及治疗进展进行综述。
遗传性骨骼肌通道病(skeletal muscle channelopathies,SMCs)是一组因骨骼肌离子通道基因突变引发的异质性遗传性疾病。根据临床表现,SMCs被分为两大类:非营养不良性肌强直(non-dystrophic myotonias,NDMs)和周期性麻痹(periodic paralyses,PPs)。NDMs包括先天性肌强直(myotonia congenital,MC)、先天性副肌强直(paramyotonia congenital,PMC)和钠通道肌强直(sodium-channel myotonia,SCM)等。PPs分为低钾型(hypokalemic periodic paralysis,hypoPP)、正常血钾型(normokalemic periodic paralysis,normoPP)、高钾型(hyperkalemic periodic paralysis,hyperPP)和Andersen-Tawil综合征(andersen-tawil syndrome,ATS)。NDMs和PPs在临床表型和分子层面上存在重叠,少数患者同时表现出非肌营养不良性肌强直和周期性麻痹表型,这进一步表明SMCs在临床谱系中是一个连续的过程。
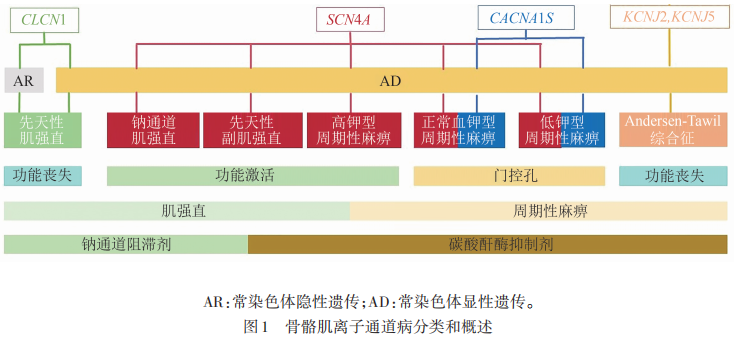
骨骼肌电压门控离子通道主要包括钙通道(Cav1.1,CACNA1S)、钠通道(Nav1.4,SCN4A)、钾通道(Kv家族,KCNJ2、KCNJ5等)、氯通道(ClC-1,CLCN1)等,它们在肌肉去极化和复极化的收缩过程中起关键作用。静息状态下,这些通道维持离子稳态,确保肌膜具备正常的应激反应能力。当骨骼肌离子通道发生突变时,肌膜通道功能受损,可能导致两种不同的病理状态:肌膜对神经刺激反应过度或持续时间延长(兴奋性增高),患者表现为肌肉过度兴奋、自主收缩后延迟放松;或者出现反应不足(兴奋性降低),患者则表现为程度不等的肌无力发作。钙通道的生理功能为介导兴奋-收缩耦联,触发肌浆网RyR释放Ca²⁺,钙通道突变可导致PPs。钠通道的生理功能是引发肌膜去极化,启动兴奋-收缩耦联,快速激活与失活,钠通道突变可导致PPs、PMC、SCM。钾通道的生理功能是复极化阶段K⁺外流终止动作电位,维持静息电位,部分亚型参与稳定静息膜电位,钾通道突变可导致ATS。氯通道的生理功能是维持膜稳定性,通过Cl⁻外流抵消K⁺外流引起的超极化,防止肌膜过度兴奋,氯通道突变可导致MC。本研究对骨骼肌离子通道病的分类、临床表现、诊断标准、基因病理分型、致病机制、治疗方法及治疗进展进行综述(见图1)。
1 周期性麻痹
原发性周期性麻痹(PPs)是最早被发现与离子通道异常有关的遗传性疾病。临床表现为发作性肌无力等特征,其发病率约为1/10万。此病多见于儿童和青年人。本团队对中国周期性麻痹队列研究发现:中国周期性麻痹患者散发为主(约80%),西方国家与此相反;发病高峰在20~40岁,较西方国家患者发病年龄(10~20岁)相对晚;男性患者比例约占97%,较西方国家(约62%)更高;携带致病突变的家系调查发现,男女携带致病变异的比率相近,但是女性携带者外显率明显降低,尤其是钠通道突变携带者。
2 临床表现
2.1 临床症状与体征
低钾型周期性麻痹(hypoPP):是最常见的原发性周期性麻痹类型,其特征性的临床表现为:(1)多在1~20岁起病,男性多于女性;(2)一般在晨起时发现四肢无力,经数小时至数日可渐渐恢复。四肢肌肉较易受累,近端重于远端;通常脑神经支配肌肉和呼吸肌不易受累;少数严重患者可出现呼吸肌瘫痪或心律失常而危及生命;(3)发作间期一般肌力正常,但有部分患者(约25%)可发展成为持久性肌无力或肌萎缩。发作频率不等,15~35岁期间发作频率最高,之后随年龄增长发作次数逐渐减少;(4)诱发因素包括:饱食、剧烈运动后,感染、创伤、情绪激动,月经、受凉等。对我国hypoPP的研究发现:与西方国家相比,我国hypoPP散发患者更为多见,发病年龄相对晚,高峰在10~30岁。
高钾型周期性麻痹(hyperPP),主要表现为发作性肌无力伴血钾升高。(1)多在10岁前起病,男性多;(2)发作常在晨起后早饭前出现四肢无力,持续时间15 min~2 h不等,多自行缓解,适当活动可以缩短发作时间;(3)发作期常伴有肌肉疼痛、僵硬感,手肌、舌肌可有肌强直发作,在面部、舌、鱼际及手指伸肌易观察到肌强直,但不影响患者的自主活动,一般不伴有心律失常和呼吸肌无力;高钾饮食、升高血钾的药物、运动后休息、饥饿、紧张、寒冷、压力及怀孕可诱发;(4)一般病初发作次数少,随着年龄的增加发作频率和严重性逐渐增加,直到约50岁后,频率开始显著减少;部分患者可发展成为持久性肌无力和肌萎缩,主要累及下肢的肌肉;(5)与hypoPP的区别除了发作时血钾不同外,还有发作短暂而频繁,饱食后不诱发,口服氯化钾症状加重。
正常血钾型周期性麻痹(normoPP):临床上主要表现为发作性肌无力伴随血清钾浓度正常。多在10岁前发病,肌无力的时间较长,往往持续数天至数周,发作期血钾及尿钾均在正常范围,限制盐的摄入或补充钾盐可诱发和加重本病,补钠后好转。
Andersen-Tawil综合征(ATS):是非常罕见的特殊类型周期性麻痹,约占周期性麻痹患者的10%。不同于其他类型的周期性麻痹,它是多系统受累离子通道病。青少年期发病多见,以周期性麻痹、室性心律失常及发育异常3种临床表现为主要临床特征。ATS可伴发低血钾、正常血钾和高血钾,以低钾型最为多见,不伴肌强直;通常发作频率和严重程度随着年龄增长而有所下降;高碳水化合物饮食、剧烈运动、紧张、寒冷、月经可诱发。心脏受累征象可表现室性心律失常,心电图可见宽大U波、Q-T间期延长、室性二联律或三联律、双向室性心动过速等。ATS的Q-T间期延长被指定为长QT综合征7(LQT7),危及生命的心律失常不像其他长QT综合征那样常见,但确实会发生,甚至无症状患者也需要定期的心脏病随访。面部和骨骼发育畸形是ATS特征表型之一。独特的面部特征可表现为眼窝凹陷、前额宽、眼距过宽、眼裂短小、低位耳、阔鼻、薄上唇、高腭弓、牙齿异常(恒牙萌出延迟和缺牙)、小下颌骨和上颌骨。骨骼异常包括手脚小、身材矮小、脊柱侧凸、手指和脚趾弯曲、第二和第三脚趾轻度并指。78%患者具有2种畸形特征,其中以先天性指(趾)弯曲和下颌发育不全最常见。
2.2 辅助检查
实验室生化检查:hypoPP发作期血清钾水平低于正常,发作间期血清钾水平正常;hyperPP在发作期血钾可高于正常水平;normoPP血钾正常;ATS发作期血清钾水平:正常、升高或降低。
心电图检查:hypoPP发作期心电图呈低钾性改变,可出现U波,S-T段下降,T波低平、增宽、双向、倒置;可出现各种心律失常,以窦性心动过速、室性早搏、阵发性心动过速为最常见;hyperPP发作期可表现T波高而尖、S-T段压低,P-R间期延长,P波增宽、低平、消失,Q-T间期延长,QRS增宽与T波融合等高血钾心电图的表现;normoPP心电图正常;ATS呈室性心律失常,心电图可见宽大U波,Q-T间期延长。
神经电生理检查:hypoPP发作期运动神经传导复合肌肉动作电位(CMAP)波幅降低。运动诱发实验是发作间期评估原发性周期性麻痹的重要电生理手段。短时运动诱发实验:常温下,小指展肌大力收缩10 s后,记录收缩前,运动后即刻,每间隔10 s~1 min记录CMAP波幅,重复3次。长时运动诱发实验:常温下,小指展肌大力收缩15 s后,放松5 s,重复至5 min后,记录收缩前,运动后即刻,每间隔5~40 min记录复合肌肉动作电位。CMAP改变百分比计算公式:(运动后波幅一运动前波幅)/运动前波幅×100%。在发病间期,低钾性周期性麻痹患者短时运动诱发实验正常。长时运动诱发实验阳性,运动后40 min内波幅/面积较基线值下降大于40%;hyperPP肌电图表现为发作期运动单位减少或无反应;在发作间期,50%的患者可以出现肌强直电位;部分患者,尤其是有持久性肌无力的患者可有肌病表现。发作间期的运动诱发试验可有助于诊断:短时运动诱发实验阳性;长时运动诱发实验阳性,患者CMAP波下降明显,尤其在运动后的前20 min内下降速度最快;normoPP肌电图表现为短时运动诱发试验CMAP升高,长时运动诱发试验阳性,同时可有肌强直电位发放;ATS长时运动诱发试验阳性。
病理检查:周期性麻痹患者虽然肌肉病理可以有异常发现,但是肌肉病理不是确诊所必需的检查。一般出现持久性肌无力患者,需要与其他肌病鉴别时,建议肌肉活检病理检测,排除其他肌病。原发性周期性麻痹肌肉病理可呈空泡样肌病表现,但没有特异性;与致病基因相关,钙通道基因突变患者肌纤维出现空泡样变,钠通道基因突变患者肌纤维出现管状聚集。
2.3 分子遗传诊断、分型、发病机制
低钾型周期性麻痹呈常染色体显性遗传,CACNA1S基因病理突变导致hypoPP Ⅰ型,SCN4A基因病理突变导致hypoPP Ⅱ型,其他为hypoPP Ⅲ型;其中,Ⅰ型约占80%,Ⅱ型约占10%,Ⅲ型约占10%。目前已发现与hypoPPⅠ型相关的CACNA1S基因病理突变20余种,其中c.1583G>A(p.R528H)和c.3659G>A(p.R1239H)是热点突变;与hypoPPⅡ型相关的SCN4A基因病理突变近20种,其中c.2015G>A(p.R672H)和c.3395G>A(p.R1132Q)是热点突变。
hypoPP致病基因CACNA1S、SCN4A基因分别编码骨骼肌L‑型电压门控钙通道α1亚单位(Cav1.1α)和骨骼肌电压门控钠通道α亚单位(Nav1.4α)。钙或钠通道的α亚单位都是由4个同源结构域Ⅰ~Ⅳ围成一个离子孔道,每个结构域均包含6个跨膜α螺旋结构S1~S6,S4片段是由反复重复的1个阳电荷残基精氨酸和2个疏水氨基酸所构成,使通道具有电压感受器的作用。位于骨骼肌电压门控钠通道α亚单位(Nav1.4α)S4区精氨酸突变的功能研究发现,突变位点产生了一个独立于正常离子通道孔的附属离子通道,称为门控孔电流(the gating pore current,Igp),该门控孔电流在静息电位时是激活的,在去极化(激活电压感受器)时关闭。在静息电位时门控孔电流携带一个质子或Na+离子流导致肌纤维去极化。目前的假说认为,在细胞外血钾浓度正常(3.5~5.5 mmol/L)时,静息电位由90 mV去极化到87.3 mV,去极化并不明显。但是,细胞外钾浓度降低到3.0 mmol/L以下(如在剧烈运动后,进食高碳水化合物饮食等外界因素诱发下),静息电位纤维去极化到60 mV,产生电压依赖的钠通道失活、弛缓性肌肉麻痹、细胞内钠超载、细胞病理学等周期性麻痹的特征。在骨骼肌L-型电压门控钙通道α1亚单位(Cav1.1α)突变鼠模型和携带骨骼肌L-型电压门控钙通道α1亚单位(Cav1.1α)突变患者的肌纤维中也记录到门控孔电流。门控孔电流的发现是hypoPP发病机制研究的一个突破,为研发有效预防其发作的药物奠定了基础。
高钾型周期性麻痹呈常染色体显性遗传,已发现与hyperPP相关的SCN4A基因病理突变30余种,其中c.2111C>T(p.T704M)和c.4774A>G(p.M1592V)是热点突变。SCN4A基因病理突变影响该通道的门控行为,产生以失活受损和活化增强为特征的异常功能获得机制。
正常血钾型周期性麻痹呈常染色体显性遗传,CACNA1S基因病理突变导致normoPPⅠ型,SCN4A基因病理突变导致normoPPⅡ型。已发现与normoPPⅠ型相关的CACNA1S基因病理突变有c.3667A>G(p.R1242G),与normoPPⅡ型相关的SCN4A基因病理突变常见有c.2023C>G(p.R675G)、c.2024G>A(p.R675Q)、c.2023C>T(p.R675W)等。功能研究发现:突变离子通道也存在一个独立于正常离子通道的附属离子通道门控孔电流,门控孔电流在激活和慢性失活状态下开放,导致离子通道功能异常。
Andersen-Tawil综合征呈常染色体显性遗传,KCNJ2基因突变导致ATSⅠ型,其他为ATSⅡ型;其中,Ⅱ型约占70%,Ⅱ型约占30%。已发现与ATS相关的KCNJ2基因病理突变30余种,常见的突变有c.199C>T(p.R67W)、c.652C>T(p.R218W)等。病理突变是功能缺失突变,使内向整流钾电流减少、细胞动作电位复极化减慢、动作电位持续时间和静息膜电位去极化延长,从而导致周期性麻痹和室性心律失常。
3 临床诊断
3.1 hypoPP临床诊断主要根据
(1)发作性四肢无力一般>2 h,发作时伴有血钾降低,补钾治疗有效;(2)诱发因素:运动后休息,碳水化合物摄入过多。排除继发性低钾血症;(3)有阳性家族史;(4)神经电生理发作间期长时运动诱发试验阳性(CMAP减少>40%);(5)基因检测证实携带CACNA1S、SCN4A基因病理突变。
3.2 hyperPP临床诊断主要根据
(1)发作性四肢无力一般<2 h,发作时伴有血钾升高;(2)诱发因素:运动后休息,钾负荷,禁食。排除继发性高钾血症;(3)有阳性家族史;(4)发作间期长时运动诱发试验阳性(CMAP减少>40%);(5)基因检测证实携带SCN4A基因病理突变。
3.3 normoPP临床诊断主要根据
(1)发作性四肢无力一般持续数天,发作时伴有血钾正常,补钾治疗无效;(2)有阳性家族史;(3)发作间期长时运动诱发试验阳性(CMAP减少>40%);(4)基因检测证实携带SCN4A、CACNA1S基因病理突变。
3.4 ATS临床诊断主要根据
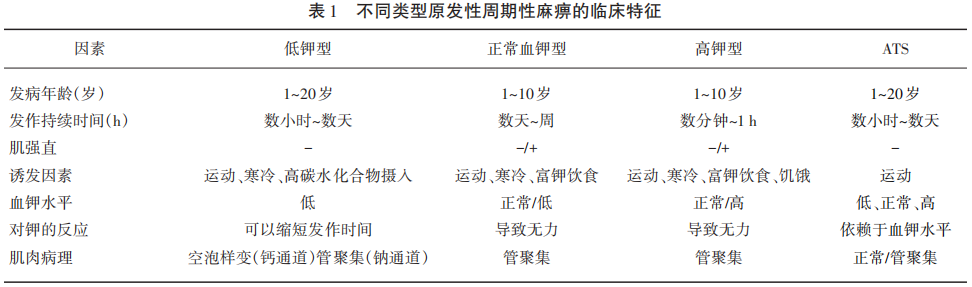
(1)发作性四肢无力一般持续数小时或数天,发作时血钾正常、偏低或升高;室性心律失常,心电图可见宽大U波,Q-T间期延长、室性二联律或三联律、双向室性心动过速;面部或骨骼发育畸形;(2)有阳性家族史;(3)发作间期长时运动诱发试验阳性(CMAP减少>40%);(4)基因检测证实携带KCNJ2基因病理突变。各型周期性麻痹鉴别诊断见表1。
4 治 疗
4.1 低钾型周期性麻痹的治疗
发作期治疗:迅速纠正低钾血症,以期缩短发作时间。首选口服钾盐,不仅有效且可避免高钾血症,首次口服10%氯化钾或枸橼酸钾30~40 ml,此后每2 h口服20 ml直到症状好转,24 h内给予钾总量可达10~15 g;疗效欠佳者可继续口服10%氯化钾或枸橼酸钾30~60 ml,直到好转。如果有呕吐,吞咽困难者可给予静脉补钾,避免使用葡萄糖盐水,因为它们可能加重肌无力,在静脉补钾过程中应作心电图和血清钾水平检测,避免发生高钾血症。
预防治疗:首先应避免诱发因素,包括高碳水化合物饮食、过劳、过饱、出汗过多、饮酒、受寒,对肾上腺素、胰岛素、激素类药物应慎用,推荐低钠、低碳水化合物和富钾饮食;发作频繁者,需要药物预防发作:(1)可长期口服氯化钾1~2 g,每日3次;(2)服用碳酸酐酶抑制剂(carbonic anhydrase inhibitors,CAIs):针对坚持补钾仍有频繁发作的患者,如乙酰唑胺(acetazolamide)、醋甲唑胺(methazolamide)、双氯非那胺(dichlorphenamide),同时应服用大量的水以预防肾结石的发生;(3)服用保钾利尿药:针对服用碳酸酐酶抑制剂无效或加重的患者,可使用保钾利尿药,如氨苯蝶啶(triamterene)、安体舒通(antisterone)。
4.2 高钾型周期性麻痹的治疗
发作期治疗:(1)通过持续温和地运动,部分患者可缓解发作;(2)静脉注射葡萄糖酸钙;(3)静脉注射葡萄糖和胰岛素;(4)沙丁胺醇(salbutamol)吸入。
预防治疗:(1)避免诱发因素:避免高钾饮食(如果汁)和药物,避免禁食、剧烈的体力劳动、暴露于寒冷环境;(2)排钾利尿剂:可服用噻嗪类利尿剂预防发作,剂量尽可能低,如双氢克尿噻25~75 mg/d,或隔日,早晨晨起服用;(3)碳酸酐酶抑制剂:乙酰唑胺、醋甲唑胺或双氯非那胺;(4)美西律(mexiletine)有利于治疗肌强直;(5)患者在需要麻醉的时候应避免使用阿片类或去极化麻醉药,例如抗胆碱酯酶、琥珀胆碱(succinylcholine)等;(6)随访和监测:患者应该定期检测肌力、监测血钾浓度。
4.3 正常血钾型周期性麻痹的治疗
发作期治疗:静脉注射葡萄糖酸钙,或大剂量生理盐水静注。
预防治疗:碳酸酐酶抑制剂,如乙酰唑胺、醋甲唑胺或双氯非那胺,保持高钠低钾饮食,防止过劳、寒冷和过热。
4.4 Andersen-Tawil综合征的治疗
发作期治疗:治疗心律失常和减少周期性麻痹的发作。对于不同类型的心律失常及血钾浓度进行相应处理。
预防治疗:需要多学科参与和管理,患者需要每年进行随访和进行长程心电图等心脏情况监测,如果有症状性心律失常,可以选用β受体阻滞剂、钠通道阻滞剂(氟卡尼)、钙通道阻滞剂、心脏起搏器植入或埋藏式心律转复除颤器(ICD)治疗。
4.5 治疗进展
2016年,有学者发现周期性麻痹患者使用辅酶Q10治疗有效,目前正在进行临床试验。建议辅酶Q10 10 mg/次,3次/d或者艾地苯醌30 mg/次、3次/d。
2014年,有学者提出Na-K-2Cl协同转运蛋白(NKCC1)抑制剂Bumetanide(布美他尼)可能对低钾型周期性麻痹的有治疗效果,布美他尼可以抑制NKCC1,从而限制与重复动作电位相关的细胞内氯浓度升高,导致在细胞外低钾状态下,肌纤维膜稳定,这有助于防止肌无力的发生和发展,并可以促进肌无力的恢复。在对NaV1.4和CaV1.1突变的hypoPP小鼠模型研究中发现,对高钾型周期性麻痹动物模型无效。之后,一项随机、双盲、安慰剂对照的布美他尼治疗hypoPP Ⅱ期临床试验研究。入组10例CACNA1S突变hypoPP患者。长时运动诱发实验中CMAP振幅下降比率作为主要评价指标。结果显示:服药后1 h,布美他尼组和安慰剂组峰值振幅的平均CMAP百分比差异无统计学意义;但与无安慰剂组相比,治疗组中有2例患者从肌无力发作中恢复过来,没有严重不良事件发生。这个临床试验是阴性结果,分析可能有以下几种原因:样本量小、肢体活动时间长、临床试验在发作期用药而非预防用药,与动物实验相比临床试验治疗剂量相对较低造成的。未来仍需进一步扩大样本量,临床研究该药物治疗hypoPP的可能性。
新型钾通道开放剂:Retigabine(瑞替加滨)是KCNQ开放剂,增强钾电流,减缓通道失活。既往主要用于成人难治性部分性癫痫发作的辅助治疗。retigabine(1~5 µm)对1型(CACNA1S突变)和2型(SCN4A突变)hypoPP小鼠模型治疗中,可有效预防低钾诱发的肌无力,并促进肌无力发作后的恢复。这个药有望今后进行临床试验加以验证。
5 非肌营养不良性肌强直
非肌营养不良性肌强直(nondystrophic myotonias,NDMs)是罕见的骨骼肌离子通道病,临床表现为自主收缩或叩击后肌肉延迟松弛,导致肌肉僵硬、疼痛、疲劳和无力等特征,发病率在0.75/10万~1.70/10万。根据临床表现及基因病理突变的类型将其分为先天性肌强直(myotonia congenital,MC)和先天性副肌强直(paramyotonia congenital,PMC),钠通道肌强直(sodium-channel myotonia,SCM)。
5.1 临床症状与体征
5.1.1 先天性肌强直
先天性肌强直(MC)是最常见的非萎缩性肌强直,患病率为0.2/10万~7.3/10万。根据遗传方式分为常染色体显性遗传的Thomsen型和常染色体隐形遗传的Becker型。
Thomsen型先天性肌强直临床特征为:(1)多在婴儿期或儿童早期发病。(2)常表现为上肢远端和颜面肌群短暂性、无痛性肌强直,肌无力和多系统损害不明显;肌肉反复收缩,引起肌肉过饱满、肥大,貌似运动员体型;肌强直不随病程而加重。(3)诱发因素包括:情绪激动、怀孕、甲状腺功能减退、麻醉剂和寒冷暴露等。
Becker型先天性肌强直临床特征为:(1)常在儿童晚期发病;发病时间较Thomsen型晚,但症状较其重。(2)常常从下肢开始发病,逐渐上升,常表现为手肌、颈肌等乃至全身骨骼肌均可发生肌强直;多数患者肌强直后有短暂肌无力,运动后可减轻;下肢明显的肌肥大,呈运动员体型,肌力正常;可有关节挛缩,患者出现足尖走路和代偿性脊柱前弯;症状进行性加重,至成人期趋于稳定。(3)诱发因素包括:寒冷、情绪、月经和怀孕等。Thomsen型和Becker型均可出现反复运动后肌强直症状减轻的加温现象(warm-up phenomenon)。
5.1.2 先天性副强直
先天性副强直(PMC)是常染色体显性遗传骨骼肌离子通道病,患病率为1/25万。临床特征为:(1)患者一般在10岁之前就发病;(2)面肌、眼肌、颈肌及上肢的肌强直,面肌及眼肌强直更为常见,部分伴双下肢轻度受累;可出现反常性肌强直(反复运动后肌强直症状加重);部分患者可伴发肌无力,持续数小时到数天,少数患者可发展成持久性肌无力;可伴肌肉疼痛;(3)诱发因素包括:寒冷、运动、妊娠、甲减、禁食等;缓解因素:保暖。
5.1.3 钠通道肌强直
钠通道肌强直(SCM)是常染色体显性遗传骨骼肌离子通道病,其发病率约为1/40万。临床上表现为一种纯粹的肌强直不伴肌无力,相关基因SCN4A,是一个谱系病,包括有多个亚型:钾加重肌强直(potassium-aggravated myotonia)、波动性肌强直(myotonia fluctuans)、持久性肌强直(myotonia permanens)、乙酰唑胺反应性肌强直(acetazolamide-responsive myotonia)、痛性肌强直(painful myotonia)等。其临床特征为:(1)患者一般在10岁之前发病。(2)面肌及眼肌强直明显,用力闭眼后睁眼困难、言语不清,肌强直可伴有肌肥大,肌痛。(3)诱发和加重因素包括:情绪波动、压力、由静止到运动、钾离子加剧肌强直;缓解因素:反复运动后肌强直症状减轻的加温现象(warm-up phenomenon)。(4)不伴肌无力。(5)对温度不敏感。
5.2 辅助检查
5.2.1 实验室生化检查
(1)Thomsen型血清肌酸激酶正常或轻度升高;(2)Becker型血清肌酸激酶轻度升高,较Thomsen型升高明显,但不超过正常上限的3~4倍;(3)PMC和SCM血清肌酸激酶可轻度升高。
5.2.2 神经电生理检查
(1)针极肌电图:可见肌强直电位,插入电位延长,扬声器发出轰炸机俯冲般或蛙鸣样声音,运动单位时限缩短,波幅下降;感觉和运动神经传导速度正常。(2)短时运动诱发实验方法:常温下,右手小指展肌大力收缩10 s后,记录收缩前,运动后即刻,每间隔10 s~1 min记录运动神经传导复合肌肉动作电位(CMAP)波幅,重复3次;低温下短时运动诱发实验:冰袋置于受试者左手7 min,使左手温度降低,重复上述短时运动诱发实验。短时运动诱发实验有3种模式:Fournier pattern Ⅰ,运动10 s后CMAP波幅和面积快速下降>20%,重复运动后CMAP波幅进一步下降;Fournier pattern Ⅱ,运动10 s后CMAP波幅和面积快速下降>20%,在1 min内快速恢复,重复运动后CMAP波幅下降幅度降低;Fournier pattern Ⅲ,运动10 s后1 min内CMAP波幅和面积无明显变化,重复运动后两者也无明显变化。一般而言,PMC表现为Fournier pattern Ⅰ,MC在室温和低温下表现为Fournier pattern Ⅱ。SMC表现为Fournier pattern Ⅲ。(3)长时运动诱发实验:常温下小指展肌大力收缩15 s后,放松5 s,重复至5 min后,记录收缩前,运动后即刻,每间隔5~40 min记录复合肌肉动作电位。MC和SMC长时运动诱发实验大致正常,PMC长时运动诱发实验阳性。
5.2.3 神经影像学检查
肌肉MRI示大部分MC患者大腿肌肉可以出现均匀一致的脂肪化,小腿肌肉有选择性脂肪化。肌肉水肿也很常见,尤其是小腿的腓内肌中央出现STIR序列高信号,呈特异性的“中央条纹”征。
5.2.4 分子遗传诊断、分型
MC是由CLCN1基因病理突变所致,可分为显性遗传Thomsen型和隐性遗传Becker型。已发现与MC相关的CLCN1基因病理突变30余种,其中c.892G>A(p.A298T)是热点突变。PMC呈常染色体显性遗传是由SCN4A基因病理突变所致,已发现与PMC相关的SCN4A基因病理突变20余种,其中c.3938C>T(p.T1313M)及c.4342C>T(p.R1448C)等是热点变异。本团队既往研究发现一个中国先天性副肌强直大家系携带SCN4A基因c.4097A>G,p.N1366S新突变,通过功能验证其损害了钠通道激活稳态,导致钠通道去极化,继发通道失活,证实其导致先天性副肌强直的分子机制。SCM呈常染色体显性遗传是由SCN4A基因病理突变所致,其中已发现与SCM相关的SCN4A基因病理突变10余种,未发现明显热点变异。本团队既往还研究发现一个中国钠通道肌强直家系携带SCN4A基因c.2108T>C,p.L703P新突变,通过功能验证其导致钠通道电流密度降低、恢复受损、激活增强和失活缓慢,证明了此突变是导致钠通道肌强直的病理性突变。
5.2.5 病理
MC患者骨骼肌无特异性病理变化。幼儿期即可出现轻度肌纤维大小不一,ⅡB肌纤维萎缩;成人后ⅡB肌纤维可缺失。PMC多数患者骨骼肌病理检测可见肌纤维大小不一,偶见管状聚集、空泡等特殊结构,肌原纤维网紊乱。
5.3 临床诊断
5.3.1 MC
根据以下临床特征和辅助检查诊断。临床表现符合肌强直伴有肌肥大,反复运动后肌强直减轻(Warm-up现象);血清肌酸激酶水平轻度升高,肌电图示肌强直电位,短时运动诱发实验Fournier pattern Ⅱ模式,肌肉MRI可见肌肉脂肪化或水肿;有阳性家族史;常染色体隐性遗传(Thomsen型)或常染色体显性遗传(Becker型)的家系特点。CLCN1基因病理突变。
5.3.2 PMC
根据以下临床特征和实验室检查诊断。临床表现符合反常性肌强直,可伴发肌肉疼痛,对温度敏感,低温可诱发发作,保暖后症状缓解;血清肌酸激酶水平轻度升高,电生理检测:短时运动诱发实验异常Fournier pattern Ⅰ模式,长时运动诱发实验异常:运动后40 min内波幅/面积较基线值下降大于40%;阳性家族史;SCN4A基因病理突变。
5.3.3 SCM
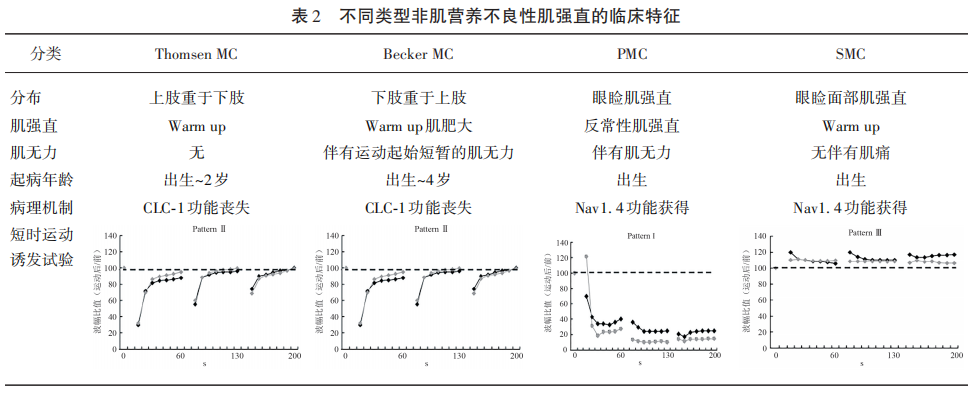
根据以下临床特征和实验室检查诊断。临床表现符合肌强直可伴有肌肥大,反复运动后肌强直减轻(Warm-up现象),对温度不敏感,不伴肌无力;肌电图示肌强直电位,短时运动诱发实验Fournier pattern Ⅲ模式,长时运动诱发实验正常;有阳性家族史;SCN4A基因病理突变。各型非肌营养不良性肌强直的临床特征鉴别见表2。
5.4 治疗
对于NDMs的管理,建议患者注意饮食和生活方式调整,避免过度劳累和受凉。肌强直症状如果影响到日常工作生活,建议进行药物干预。
临床研究显示,美西律(mexiletine)是首选抗肌强直药物,对各种类型非肌营养不良性肌强直均有效(ClinicalTrials.gov Identifier:NCT00832000)。美西律是Ib类抗心律失常电压门控钠通道阻滞剂,它的主要作用是通过增强钠通道的快速失活起效。服药过程中需注意美西律对心率的影响。若患者不能耐受美西律,可选择抗癫痫药物拉莫三嗪(lamotrigine)、卡马西平(carbamazepine),Ic类抗心律失常药物钠通道阻滞剂氟卡尼等。碳酸酐酶抑制剂如乙酰唑胺(acetazolamide)、醋甲唑胺(methazolamide)或双氯非那胺(diclofenamide)对部分MC和PMC患者有效。尤其对于寒冷加重的PMC,既可以改善肌强直也可以改善肌无力症状。
琥珀胆碱(succinylcholine)、肾上腺素(epinephrine)、β肾上腺素能受体激动剂(β-adrenergic agonists)、心得安(propranlolum)和秋水仙碱(colchicine)可能会加重病情,应避免使用。
6 总结与展望
在NDMs治疗领域,已有实验室研究显示沙芬酰胺(safinamide)作为一种神经元钠通道阻滞剂,它对肌强直的治疗有一定的潜力。研究发现,利鲁唑(riluzole)抑制持续性钠电流,也显示出抗肌强直价值,但这些药物的疗效需要进一步的临床试验加以验证。针对KCNQ通道的K+通道激活剂-瑞替加滨在治疗癫痫中是有效的,因为它引起神经元中静息膜电位的超极化。在一项研究中,研究者发现瑞替加滨在体外显著减少了肌强直的持续时间和肌强直的严重程度,对其作用机制的详细研究表明,瑞替加宾对肌肉兴奋性、静息电位、输入阻抗或单个动作电位没有影响,它可能是通过在动作电位中激活K+电流来缩短肌强直。瑞替加宾对肌肉本身的显著效果表明,激活钾电流可能是治疗肌肉过度兴奋障碍的有效方法之一,后续需要临床试验验证。
综上,离子通道病的诊疗已从传统的经验治疗逐步转向基于基因诊断和分子机制的精准医疗。未来的研究方向包括:开发更有效的药物,如针对特定离子通道突变的靶向治疗。基因治疗技术的应用,为骨骼肌离子通道病患者提供根治可能。加强对骨骼肌离子通道病患者的早期诊断,多学科个体化管理,有望进一步改善患者的预后。
参考文献
[1]Vicino A,Brugnoni R,Maggi L. Diagnostics in skeletal muscle channelopathies[J]. Expert Rev Mol Diagn,2023,23(12):1175-1193.
[2]Matthews E,Holmes S,Fialho D.Skeletal muscle channelopathies:a guide to diagnosis and management[J]. Pract Neurol,2021,21(3):196-204.
[3]Phillips L,Trivedi JR. Skeletal muscle channelopathies[J]. Neurotherapeutics,2018,15(4):954-965.
[4]Flucher BE. Skeletal muscle CaV1.1 channelopathies[J]. Pflügers Arch,2020,472(7):739-754.
[5]Cannon SC. Sodium channelopathies of skeletal muscle[J]. Handb Exp Pharmacol,2018,246:309-330.
[6]Goslinga JA,PtáČek LJ,Tawil R,et al. Andersen-Tawil syndrome[J]. Handb Clin Neurol,2024,203:59-67.
[7]Poroca DR,Pelis RM,Chappe VM. ClC channels and transporters:structure,physiological functions,and implications in human chloride channelopathies[J]. Front Pharmacol,2017,8:151.
[8]Statland JM,Fontaine B,Hanna MG,et al. Review of the diagnosis and treatment of periodic paralysis[J]. Muscle Nerve,2018,57(4):522-530.
[9]Fialho D,Griggs RC,Matthews E. Periodic paralysis[A]//Neurogenetics [M].Part Ⅱ. Amsterdam:Elsevier,2018:505-520.
[10]陆国辉,张学.产前遗传病诊断[M].2版.广州:广东科技出版社,2020.
[11]涂 柳,曾文双,褚晓凡,等.以反复周期性麻痹首诊神经内科的原发性干燥综合征1例报告[J].中风与神经疾病杂志,2020,37(5):451-452.
[12]Ke Q,Luo B,Qi M,et al. Gender differences in penetrance and phenotype in hypokalemic periodic paralysis[J]. Muscle Nerve,2013,47(1):41-45.
[13]Vicart S,Sternberg D,Fournier E,et al. New mutations of SCN4A cause a potassium-sensitive normokalemic periodic paralysis[J]. Neurology,2004,63(11):2120-2127.
[14]Fan C,Lehmann-Horn F,Weber MA,et al. Transient compartment-like syndrome and normokalaemic periodic paralysis due to a Ca(v)1.1 mutation[J]. Brain,2013,136(Pt 12):3775-3786.
[15]Sansone V,Tawil R. Management and treatment of Andersen-tawil syndrome (ATS)[J]. Neurotherapeutics,2007,4(2):233-237.
[16]Fournier E,Arzel M,Sternberg D,et al. Electromyography guides toward subgroups of mutations in muscle channelopathies[J]. Ann Neurol,2004,56(5):650-661.
[17]Tan SV,Matthews E,Barber M,et al. Refined exercise testing can aid DNA-based diagnosis in muscle channelopathies[J]. Ann Neurol,2011,69(2):328-340.
[18]Matthews E,Labrum R,Sweeney MG,et al. Voltage sensor charge loss accounts for most cases of hypokalemic periodic paralysis[J]. Neurology,2009,72(18):1544-1547.
[19]Jiang D,Gamal El-Din TM,Ing C,Lu P,Pomès R,Zheng N,Catterall WA. Structural basis for gating pore current in periodic paralysis[J]. Nature,2018,557(7706):590-594.
[20]Ke Q,He F,Lu L,et al. The R900S mutation in CACNA1S associated with hypokalemic periodic paralysis[J]. Neuromuscul Disord,2015,25(12):955-958.
[21]Matthews E,Portaro S,Ke Q,et al. Acetazolamide efficacy in hypokalemic periodic paralysis and the predictive role of genotype[J]. Neurology,2011,77(22):1960-1964.
[22]Sansone VA,Johnson NE,Hanna MG,et al. Long-term efficacy and safety of dichlorphenamide for treatment of primary periodic paralysis[J]. Muscle Nerve,2021,64(3):342-346.
[23]Maffè S,Paffoni P,Bergamasco L,et al. Therapeutic management of ventricular arrhythmias in Andersen-Tawil syndrome[J]. J Electrocardiol,2020,58:37-42.
[24]Rujirachun P,Junyavoraluk A,Pithukpakorn M,et al. Successful treatment of arrhythmia with β-blocker and flecainide combination in pregnant patients with Andersen-Tawil syndrome:a case report and literature review[J]. Ann Noninvasive Electrocardiol,2021,26(3):e12798.
[25]Desaphy JF,Altamura C,Vicart S,et al. Targeted therapies for skeletal muscle ion channelopathies:systematic review and steps towards precision medicine[J]. J Neuromuscul Dis,2021,8(3):357-381.
[26]Da Y,Lei L,Jurkat-Rott K,et al. Successful treatment of periodic paralysis with coenzyme Q10:two case reports[J]. Acta Myol,2016,35(2):107-108.
[27]Wu F,Mi W,Cannon SC. Beneficial effects of bumetanide in a CaV1.1-R528H mouse model of hypokalaemic periodic paralysis[J]. Brain,2013,136(Pt 12):3766-3774.
[28]Wu F,Mi W,Cannon SC. Bumetanide prevents transient decreases in muscle force in murine hypokalemic periodic paralysis[J]. Neurology,2013,80(12):1110-1116.
[29]Cannon SC. Periodic paralysis[J]. Handb Clin Neurol,2024,203:39-58.
[30]Quiñonez M,DiFranco M,Wu F,et al. Retigabine suppresses loss of force in mouse models of hypokalaemic periodic paralysis[J]. Brain,2023,146(4):1554-1560.
[31]Stunnenberg BC,LoRusso S,Arnold WD,et al. Guidelines on clinical presentation and management of nondystrophic myotonias[J]. Muscle Nerve,2020,62(4):430-444.
[32]Morales F,Pusch M. An up-to-date overview of the complexity of genotype-phenotype relationships in myotonic channelopathies[J]. Front Neurol,2020,10:1404.
[33]Jeng CJ,Fu SJ,You CY,et al. Defective gating and proteostasis of human ClC-1 chloride channel:molecular pathophysiology of myotonia congenita[J]. Front Neurol,2020,11:76.
[34]Morrow JM,Matthews E,Raja Rayan DL,et al. Muscle MRI reveals distinct abnormalities in genetically proven non-dystrophic myotonias[J]. Neuromuscul Disord,2013,23(8):637-646.
[35]Ke Q,Ye J,Tang S,et al.N1366S mutation of human skeletal muscle sodium channel causes paramyotonia congenita[J].J Physiol,2017,595(22):6837-6850.
[36]Ke Q,Zhao Y,Li Y,et al. Clinical comparison and functional study of the L703P:a recurrent mutation in human SCN4A that causes sodium channel myotonia[J]. Neuromuscul Disord,2022,32(10):811-819.
[37]Vivekanandam V,Skorupinska I,Jayaseelan DL,et al. Mexiletine versus lamotrigine in non-dystrophic myotonias:a randomised,double-blind,head-to-head,crossover,non-inferiority,phase 3 trial[J]. Lancet Neurol,2024,23(10):1004-1012.
[38]Desaphy JF,Modoni A,Lomonaco M,et al. Dramatic improvement of myotonia permanens with flecainide:a two-case report of a possible bench-to-bedside pharmacogenetics strategy[J]. Eur J Clin Pharmacol,2013,69(4):1037-1039.
[39]Dupont C,Denman KS,Hawash AA,et al. Treatment of myotonia congenita with retigabine in mice[J]. Exp Neurol,2019,315:52-59.
- 搜索
-
- 1000℃Nutrients:真实世界数据,纤维肌痛患者的饮食与运动自适应规律
- 1000℃D-二聚体升高诊治与管理专家共识(2026)
- 1000℃专家论坛|文良志:门静脉血栓的诊断和治疗
- 1000℃首例儿童NF2驱动型胸膜间皮瘤,多方法学检测锁定NF2双等位基因失活和14/22号染色体缺失,提示与成人胸膜间皮瘤不同
- 1000℃打破误区:干扰素追求CHB功能性治愈,HBsAg为何“不降反增”?
- 1000℃迷惑性极强的肝内病灶!影像表现疑点重重,最终病理竟查出两种不同肝脏恶性肿瘤
- 1000℃指南共识|原发性肝癌分子靶向药物相关蛋白尿中西医结合诊疗专家共识
- 1000℃Diabetologia:意大利北部社区 1~100 岁人群胰岛自身抗体与乳糜泻 TGA-IgA 的年龄分布及检测方法学验证
- 精J Child Psychol Psychiatry:12种罕见神经发育障碍儿童沟通能力谱系
- 精研究发现:爱吃辣的人,心血管病和癌症死亡风险都会显著降低
- 精Nursing in Critical Care:别再指责护士了!ICU 里被遗漏的护理,根源在系统而非个人
- 精Acta Obstet Gynecol Scand:罕见病女性的妊娠并发症与母婴结局,一项单中心434种罕见病的回顾性队列研究
- 精【爱儿小醉】儿科患者术前对流层臭氧暴露与围手术期呼吸系统不良事件之间的关系:一项单中心回顾性队列研究
- 精eBioMedicine:牙龈下微生物组与脑健康存在连续关联梯度,牙周炎或成认知衰退可干预靶点
- 精军事医学研究院《自然·通讯》:自适应IrPtCu纳米酶水凝胶实现耐药菌感染伤口序贯治疗
- 精能够逆转萎缩性胃炎的两个中成药,该怎么选择?
- 荐同时性多发性原发性肺癌,左右病灶分别为EGFR和ALK阳性,考虑淋巴结肿大仅局限左肺门及血浆EGFR阳性,采用奥希替尼联合化疗
- 荐40岁女性同时罹患卵巢支持细胞-间质细胞瘤和透明细胞乳头状肾肿瘤,WES等基因检测竟为阴性
- 荐椎管内麻醉使用止血药突发气道痉挛的抢救流程解析
- 荐女子肝区无任何不适,影像提示复杂囊性病变,层层鉴别后锁定罕见胆管源性囊性肿瘤
- 荐“绘”真报告 | 病理考虑为中枢神经细胞瘤,检出脑室外神经细胞瘤的特征性变异FGFR1-TACC1融合,辅助鉴别诊断
- 荐8岁女童出现男性化症状,竟是形似「性索-间质肿瘤」的卵巢「无性细胞瘤」所致,少见KRAS/CDK4共扩增或与侵袭性有关
- 荐17例病例分析揭示常见于中年人的色素性室管膜瘤临床特征与预后,分子检测可助力临床精准诊疗
- 荐Lancet Oncol:结直肠癌腹膜转移,围手术期化疗并非必选项
- 标签列表
-
- 星座 (702)
- 孩子 (526)
- 恋爱 (505)
- 婴儿车 (390)
- 宝宝 (328)
- 狮子座 (313)
- 金牛座 (313)
- 摩羯座 (302)
- 白羊座 (301)
- 天蝎座 (294)
- 巨蟹座 (289)
- 双子座 (289)
- 处女座 (285)
- 天秤座 (276)
- 双鱼座 (268)
- 婴儿 (265)
- 水瓶座 (260)
- 射手座 (239)
- 不完美妈妈 (173)
- 跳槽那些事儿 (168)
- baby (140)
- 女婴 (132)
- 生肖 (129)
- 女儿 (129)
- 民警 (127)
- 狮子 (105)
- NBA (101)
- 家长 (97)
- 怀孕 (95)
- 儿童 (93)
- 交警 (89)
- 孕妇 (77)
- 儿子 (75)
- Angelababy (74)
- 父母 (74)
- 幼儿园 (73)
- 医院 (69)
- 童车 (66)
- 女子 (60)
- 郑州 (58)