首页 > 医疗资讯/ 正文
[摘要] 背景与目的:原发性肾上腺血管内大B细胞淋巴瘤(intravascular large B-cell lymphoma,IVLBCL)是一种罕见的高侵袭性淋巴瘤,目前对其仍缺乏充分认识。本研究旨在探讨肾上腺IVLBCL的临床病理学特征、分子遗传学特征及预后。方法:收集复旦大学附属肿瘤医院病理科2012—2023年诊断的肾上腺IVLBCL病例,回顾性分析其临床及组织病理学特征、免疫表型、治疗和预后,并采用靶向二代测序(next-generation sequencing,NGS)技术探讨其分子遗传学特点。本研究已通过复旦大学附属肿瘤医院伦理委员会审查(伦理编号:050432-4-2307E)。结果:5例肾上腺IVLBCL患者均为男性,中位年龄为52岁(年龄范围50~82岁)。2例因低热而就诊,1例表现为腹痛,1例为体检发现,1例未获得相关信息。外周血检查示2例有血清乳酸脱氢酶升高,2例出现肾上腺功能异常。影像学检查显示,肾上腺增大或占位性病变伴氟代脱氧葡萄糖(fluorodeoxyglucose,FDG)摄取增高;其中4例累及双侧肾上腺,1例累及右侧肾上腺。显微镜下可见异型大淋巴细胞局限于血管腔内,部分病例血管腔内见纤维素性坏死。免疫组织化学染色显示,所有(5/5)病例的肿瘤细胞CD20阳性,Ki-67增殖指数均较高(>80%),80%(4/5)病例呈非生发中心B细胞样(non-germinal-center B-cell-like,non-GCB)表型,100%(4/4)呈MYC/BCL2双表达;血管内皮细胞标志物染色显示,肿瘤细胞绝大多数位于血管内。3例患者获得治疗和随访信息,其中1例仅接受手术切除的患者确诊后5个月死亡,1例经手术切除后接受R-CHOP方案治疗的患者获得完全缓解,1例穿刺活检确诊后接受R-CHOP方案治疗的患者获得部分缓解;1年总生存率为66.7%,总生存期为5~87个月。1例行NGS检测,结果显示,MYD88 V217F、TP53、CDH1、ARID1B、MSH3、MLH3、PTPRK、CD22和FLCN等基因错义突变。结论:肾上腺IVLBCL发病率低,好发于中老年男性,本组病例以non-GCB为主且常伴MYC/BCL2双表达,并存在MYD88非L265P突变。肾上腺IVLBCL由于其临床症状多样化且缺乏特异性导致早期确诊困难,积累更多病例深入了解其临床病理学特征及分子遗传学特征,有助于早期诊断、及时治疗并改善患者预后,为深入认识疾病发生、发展机制、探索治疗靶点提供理论基础。
[关键词] 肾上腺;血管内大B细胞淋巴瘤;临床病理学特征;MYD88
[Abstract] Background and purpose: Primary adrenal intravascular large B-cell lymphoma (IVLBCL) is rare and highly aggressive. Unfortunately, comprehensive and sufficient understanding of the disease is lacking. This study investigated the clinicopathological and molecular genetic characteristics of adrenal IVLBCL. Methods: Adrenal IVLBCL cases diagnosed from 2012 to 2023 were collected from Department of Pathology, Fudan University Shanghai Cancer Center. The clinical and histopathological features, immunophenotype, treatment and prognosis were analyzed. The molecular genetic characteristics were detected using next-generation sequencing (NGS). This study was approved by the Ethics Committee of Fudan University Shanghai Cancer Center (Ethics number: 050432-4-2307E). Results: All of the 5 patients were male, with median age 52 years (ranged 50-82 years). Two cases had low-grade fever, 1 case had abdominal pain, 1 case was found by physical examination, and the information of the remaining one was unknown. Peripheral blood test showed elevated serum lactate dehydrogenase in 2 cases and adrenal dysfunction in 2 cases. On initial diagnosis, imaging tests displayed adrenal enlargement or masses with increased fluorodeoxyglucose (FDG) uptake. Bilateral adrenal glands were involved in 4 cases and only the right adrenal gland was involved in the other case. Morphologically, large atypical lymphocytes were confined to blood vessels, and fibrinous necrosis was observed in some cases. Immunohistochemical study revealed that CD20 was positive in all cases. Ki-67 proliferation index was high, all above 80%. 80% (4/5) of the cases were of non-germinal-center B-cell-like (non-GCB) phenotype, 100% (4/4) of the cases had MYC/BCL2 double expression. Endothelial cell markers staining indicated that most of the tumor cells were confined within the blood vessels in all cases. Follow-up data were available in 3 patients. One patient who underwent only surgical rep died 5 months after diagnosis, one achieved complete remission after surgery plus R-CHOP, and the other diagnosed by biopsy achieved a partial remission after R-CHOP. The 1-year overall survival rate was 66.7%, and overall survival was 5-87 months. NGS testing in 1 case showed missense mutations in MYD88 V217F, TP53, CDH1, ARID1B, MSH3, MLH3, PTPRK, CD22 and FLCN. Conclusion: Adrenal IVLBCL is rare and tends to occur in the middle-aged and elderly male. The majority of our patients were non-GCB phenotype, often accompanied by MYC/BCL2 double expression, and MYD88 non-L265P mutation was detected. Early diagnosis of adrenal IVLBCL is difficult due to its diverse clinical symptoms and lack of specificity. It is of great importance to accumulate more cases and further understand the clinicopathological and molecular genetic characteristics of this rare disease, which might not only help with early diagnosis, timely treatment and improvement of prognosis, but also provide a theoretical basis for further understanding the pathogenesis and development of the disease and exploring therapeutic targets.
[Key words] Adrenal gland; Intravascular large B-cell lymphoma; Clinicopathological features; MYD88
血管内大B细胞淋巴瘤(intravascular large B-cell lymphoma,IVLBCL)是弥漫性大B细胞淋巴瘤(diffuse large B cell lymphoma,DLBCL)的一种特殊亚型,特征性表现为肿瘤细胞在血管腔内选择性生长。几乎全身各个器官均可受累,包括中枢神经系统、皮肤、骨髓和肺等,其中以中枢神经系统和皮肤最为常见[1]。原发性肾上腺IVLBCL极为罕见,目前对于其临床病理学特征、治疗及预后等罕见报道。深入了解其临床病理学特征及分子遗传学特征,对于早期准确诊断、及时合理治疗、探索治疗靶点及改善患者预后等均具有重要意义。
1 资料和方法
1.1 临床材料
收集复旦大学附属肿瘤医院病理科2012—2023年诊断的肾上腺IVLBCL病例。由2名经验丰富的高年资病理科医师(从事淋巴瘤亚专科工作10年以上)复核病理学诊断,诊断标准参考第5版世界卫生组织(World Health Organization,WHO)造血与淋巴组织肿瘤分类[2]。通过查阅电子病例档案及电话随访完善临床资料及预后信息,包括年龄、性别、临床表现、影像学及实验室检查、治疗及生存情况等。本研究已通过复旦大学附属肿瘤医院伦理委员会审查(伦理编号:050432-4-2307E)。
1.2 免疫组织化学
免疫组织化学染色在BenchMark XT全自动免疫组织化学染色仪(购自美国Roche Ventana公司)中进行。一抗包括:CD20、PAX5、BCL2、CD3、CD5、CD30和Ki-67,均购自美国Roche公司;MUM1和CD31,均购自丹麦Dako公司;CD10和CD34,均购自福州迈新生物技术开发有限公司;BCL6,购自德国Leica公司;MYC,购自英国Abcam公司。结果判读标准:PAX5、BCL6、MUM1、MYC、Ki-67阳性表现为细胞核着色,CD3、BCL2和CD30阳性表现为细胞膜/细胞质着色,CD20、CD10、CD5、CD31和CD34阳性表现为细胞膜着色。CD10、 BCL6、MUM1、CD5阳性判定标准为超过30%肿瘤细胞染色,≥40%肿瘤细胞表达MYC蛋白判读为阳性,≥50%肿瘤细胞表达BCL2判读为阳性[2-3]。CD30阳性判读目前尚无统一标准,本研究参考Wawire、Campuzano-Zuluaga等的研究[4-5]将≥20%肿瘤细胞染色定义为阳性。
依据Hans分型法则[3]进行细胞起源分型,分为生发中心B细胞样(germinal-center B-cell-like,GCB)型和非GCB(non-GCB)型。依据WHO分类中的定义,将肿瘤细胞MYC蛋白表达≥40%且BCL2蛋白表达≥50%定义为双表达[2]。
1.3 靶向二代测序(next-generation sequencing,NGS)
使用QIAamp DNA Mini Kit提取基因组DNA,利用多重聚合酶链反应扩增目标区域,制备文库DNA。经文库构建、扩增,通过NEXT550进行测序。突变数据处理过程如下:① 测序原始数据BCL文件经bcl2fastq(v2.20.0)转化成FASTQ可读格式序列;② BWA(version 0.7.12)将可读序列映射至人类参考基因组( hg19)并生成SAM文件;③ sambamba (v0.8.0)将SAM文件转为压缩BAM文件;④ Picard(version 2.9.4)根据染色体坐标对其进行排序和去重;⑤ GATK(version 3.4.0)获取突变信息。以AF>5%且pop_freq≤0.001为标准筛选数据。
2 结 果
2.1 临床特征
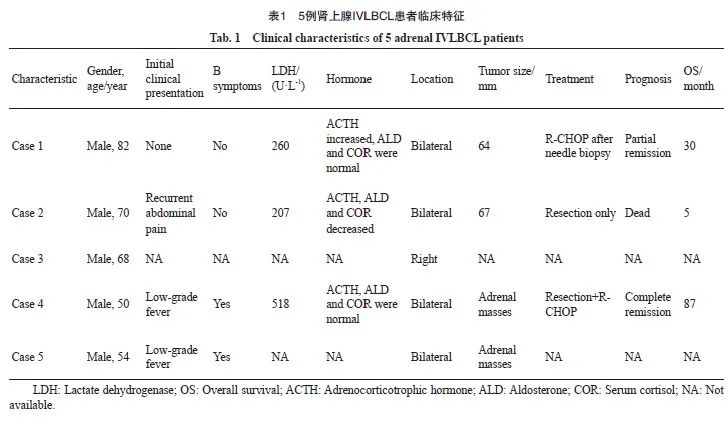
复旦大学附属肿瘤医院病理科2012—2023年诊断的DLBCL病例共9 643例,其中IVLBCL 15例,占DLBCL的0.16%;肾上腺IVLBCL仅5例,占所有IVLBCL的33.3%。本组肾上腺IVLBCL病例临床特征见表1。5例患者均为男性,中位年龄为52岁(年龄范围50~82岁)。3例为复旦大学附属肿瘤医院手术切除或穿刺活检标本;2例为外院会诊病例,均为穿刺活检标本。4例(4/5,80%)患者双侧肾上腺受累,1例(1/5,20%)仅累及右侧肾上腺。初诊时,2例(2/4,50%)表现为低热,1例(1/4,25%)因腹部胀痛而就诊,1例(1/4,25%)为体检时发现,余1例未获得相关信息。实验室检查显示,2例(2/3,67%)患者伴有血清乳酸脱氢酶升高,2例(2/3,67%)出现促肾上腺皮质激素、醛固酮或皮质醇水平异常。影像学上均表现为肾上腺增大或占位性病变伴氟代脱氧葡萄糖(fluorodeoxyglucose,FDG)摄取增高,未提示其他部位受累。
2.2 组织学形态
显微镜下肿瘤细胞呈巢团状分布(图1),细胞有明显异型性,细胞核大、圆形或不规则,部分细胞核呈空泡状、免疫母细胞或中心母细胞样,核分裂象易见。其中1例可见部分细胞核呈间变特征。4例肿瘤细胞主要位于中小血管腔内,另1例可见肿瘤细胞在大血管腔内增生聚集,伴成簇或散在分布的红细胞。部分病例血管腔内见纤维素性坏死。
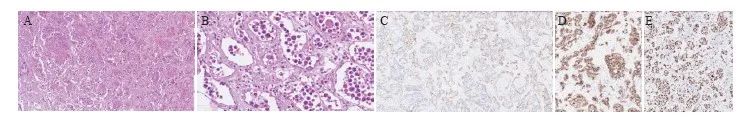
图1 肾上腺IVLBCL形态学及免疫组织化学特征
Fig. 1 Morphological and immunohistochemical features of adrenal IVLBCL
A: Atypical lymphocytes were infiltrated in sheets or clusters (H-E, ×100); B: Atypical lymphoid cells were mainly large-sized with round or oval nuclei (H-E, ×400); C: CD34 highlighted the atypical cells confined to the vascular lumina (×100); D: The neoplastic cells stained positive for CD20 (×100); E: The Ki-67 proliferation index of neoplastic cells was very high (×100).
2.3 免疫组织化学染色结果
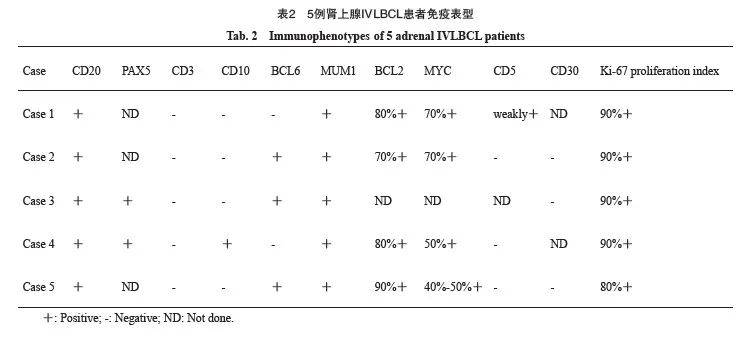
所有病例的肿瘤细胞弥漫表达广谱B细胞标记物CD20,T细胞相关抗原CD3阴性。CD10、BCL6和MUM1的阳性率分别为20%(1/5)、60%(3/5)和100%(5/5)。4例(80%)呈non-GCB表型,1例(20%)呈GCB表型。所有接受检查的病例(4/4,100%)均呈MYC/BCL2双表达。仅1例(1/4,25%)CD5表达呈弱阳性。所有病例的Ki-67增殖指数均较高(80%~90%)。血管内皮细胞标志物(CD31和CD34)染色显示,肿瘤细胞大部分位于血管腔内,仅1例在血管腔外可见小灶肿瘤细胞(表2)。
2.4 NGS分析
5例患者中,仅1例可获得足够量的石蜡包埋肿瘤组织进行NGS分析,结果显示,MYD88、TP53、CDH1、ARID1B、MSH3、MLH3、PTPRK、CD22和FLCN等基因存在错义突变。其中,MYD88为V217F突变。
2.5 治疗和预后
3例患者获得治疗和预后信息。1例患者仅接受手术切除,未行任何辅助治疗,确诊后5个月死亡;1例经手术切除后接受R-CHOP方案治疗的患者获得完全缓解;1例穿刺活检确诊后接受R-CHOP方案治疗的患者获得部分缓解。1年总生存率为66.7%,总生存期为5~87个月(表1)。
3 讨 论
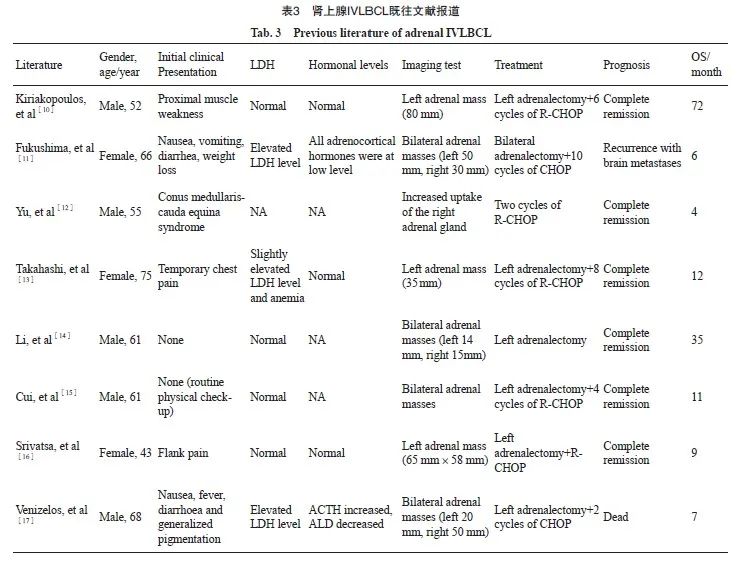
2008年第4版WHO分类[6]将IVLBCL作为DLBCL的一个独立亚型纳入淋巴瘤分类。不同结外器官和组织均可受累,以中枢神经系统和皮肤多见,病变可累及多个器官,其中2%~33%有肾上腺受累[1,7-9]。但是原发性肾上腺IVLBCL极其罕见,目前文献报道极少且多为个案报道(表3),对其临床病理学特征仍缺乏充分认识。因此,增加对这组特殊病例的临床病理学特征的认识,对于准确诊断、优化治疗策略及预后预测都具有重要意义。
肾上腺IVLBCL好发于中老年人,既往研究[1,7-9]报道,发病的中位年龄为61岁,与IVLBCL相似(64~70岁);本组肾上腺IVLBCL患者较为年轻(中位年龄为52岁)。本组患者均为男性,既往文献报道的8例肾上腺IVLBCL患者男女比例约为1.7∶1.0。与IVLBCL相似,肾上腺IVLBCL首发症状多样化,部分患者表现为非特异性消化道症状、胸痛或腰痛,部分患者无症状,少数表现为全身色素沉着。其临床表现的多样化及非特异性导致早期确诊困难,甚至易误诊。实验室检查显示,部分患者出现血清乳酸脱氢酶水平升高、贫血或肾上腺素水平异常。影像学检查显示,肾上腺病变最大径为1.4~8.0 cm。有研究[10-11,18]总结双侧肾上腺皮质肿大/占位性病变和(或)肾上腺皮质功能减退可能是肾上腺IVLBCL较常见的首发表现。因此,对于影像学上表现为肾上腺孤立性肿块/占位性病变和(或)快速进展性肾上腺功能衰竭的中老年患者,需考虑到IVLBCL的可能。影像学检查尤其是正电子发射计算机体层成像(positron emission tomography and computed tomography,PET/CT)有助于鉴别是否为其他部位DLBCL累及肾上腺[14,19]。
组织病理学特征方面,肾上腺IVLBCL与发生在其他部位的IVLBCL相似,特征性表现为肿瘤性淋巴细胞选择性在小或中等大小血管内生长[1-2,7-9],有时可以看到残留的肾上腺组织。肿瘤细胞缺乏黏附性,细胞核大深染,圆形/类圆形或不规则,核分裂象易见。血管腔内常可见纤维素样坏死。有时纤维素样坏死明显而肿瘤细胞稀少,且常伴有较多红细胞,需要与血栓、血管炎性疾病等良性病变鉴别。CD34和CD31免疫组织化学染色显示,肿瘤细胞位于血管腔内,有助于鉴别肾上腺DLBCL。有研究[7,20]显示,IVLBCL多呈non-GCB表型,40%存在MYC/BCL2双表达,且双表达患者的死亡率显著高于非双表达患者[21]。研究[8-9,20,22]报道的IVLBCL的CD5阳性率在22%~52%不等。尽管普遍认为原发性CD5阳性DLBCL患者预后比CD5阴性患者差[23],但目前有限的研究[7,9]显示, CD5阳性和CD5阴性IVLBCL患者预后没有显著差异。约44.4%的IVLBCL病例表达PD-L1[24],提示免疫治疗可能具有潜在意义。目前关于肾上腺IVLBCL免疫表型特征尚未见总结报道,本组病例免疫表型与既往文献报道的IVLBCL相似,大部分为non-GCB表型,但MYC/BCL2双表达率高于Boonsakan等[21]报道的IVLBCL,且仅1例呈CD5弱阳性。由于肾上腺IVLBCL发病率极低、文献报道少,因此需要积累更多病例进一步总结其免疫表型特征及其临床病理学相关性。
2018年有研究[20,25]相继显示IVLBCL中存在MYD88 L265P突变(44.0%~55.6%)和CD79B Y196突变(26.0%~66.7%)。Shimada等[26]研究发现IVLBCL中的突变基因在B细胞受体/核因子-κB(nuclear factor-κB,NF-κB)信号通路中显著富集,包括CD79B(67%)、MYD88(57%)和IRF4(38%)等;同时38%的IVLBCL中存在程序性死亡蛋白配体-1(programmed death ligand-1,PD-L1)/PD-L2重排。上述基因变异为IVLBCL靶向治疗或免疫治疗的选择提供了理论依据[27]。尽管Cui等[15]研究发现肾上腺IVLBCL中存在IGH/BCL6基因融合,然而目前对于肾上腺IVLBCL的分子遗传学特征所知甚少,其基因突变谱尚未见报道。本组病例中仅1例有足够量的石蜡包埋肿瘤组织可进行NGS检测,结果显示,存在MYD88、TP53、CDH1、 ARID1B、MSH3、MLH3、PTPRK、CD22和FLCN等基因突变。值得注意的是,该病例中MYD88突变并非L265P高频突变,而是V217F,后者系致病性变异[28-29]。Xie等[28]研究发现MYD88非L265P突变DLBCL患者的无进展生存期显著优于L265P突变患者。但由于目前报道较少,MYD88非L265P突变与肾上腺IVLBCL患者临床病理学特征的相关性有待进一步探讨。
除TP53突变在DLBCL中常见且提示预后不良[30]之外,个别报道胃原发DLBCL中存在CDH1突变,可能参与疾病的发生、发展[31]。ARID1B突变在DLBCL、滤泡性淋巴瘤中有报道但临床意义不明[32]。一项包含22例DLBCL患者的研究[33]显示,DLBCL中存在MSH3和MLH3基因胚系突变(分别为13.6%和9.1%),可能与疾病进展和治疗反应有关。PTPRK、CD22及FLCN基因突变目前在DLBCL中未见相关报道。肾上腺IVLBCL的分子遗传学特征以及上述基因突变在该疾病中的意义还需要积累更多病例进行探讨,以期进一步了解肾上腺IVLBCL的发病机制,并探索治疗及预后预测靶点。
IVLBCL对化疗反应较敏感,因此早期诊断和及早治疗有利于改善患者预后[1]。目前IVLBCL一般采用R-CHOP方案为基础的联合化疗,接受含利妥昔单抗治疗的IVLBCL患者总生存期和无进展生存期明显优于接受其他化疗方案的患者[1]。针对IVLBCL患者中存在的MYD88、CD79B高频突变[9],R-CHOP方案联合BTK抑制剂可能是IVLBCL治疗的新方向。近期一项前瞻性研究[34]显示,泽布替尼联合R-CHOP治疗初诊IVLBCL患者可获得较高的缓解率(9例入组患者的总缓解率为100%),且无复发(中位随访时间为10个月)。总体而言,IVLBCL患者预后较差,中位总生存期为11~105个月[1,7-9],1年总生存率为42.3%~66.2%[1,7-8,35],3年总生存率为11.5%~51.8%[1,5,7-8,29]。本组肾上腺IVLBCL病例的1年总生存率与之接近,但病例数较少,关于其预后分析仍需要积累更多病例进一步研究,以期为临床治疗选择及预后预测等提供依据。
综上所述,肾上腺IVLBCL极为罕见,好发于中老年男性,由于缺乏特异性临床表现导致早期诊断困难,总体预后较差。对于影像学上表现为肾上腺孤立性肿块或占位性病变的中老年患者,鉴别诊断需考虑到IVLBCL。肾上腺IVLBCL多为non-GCB表型,伴MYC/BCL2双表达,并存在MYD88突变。熟悉其临床病理学特征,有助于准确诊断、早期干预,从而改善患者预后。积累更多病例深入了解其分子遗传学特征,对于理解疾病发病机制、探索治疗和预后预测靶点并精准治疗均具有重要意义。
利益冲突声明:所有作者均声明不存在利益冲突。
作者贡献声明:
林佳鑫:收集数据,撰写文章;
魏然:数据分析整理;
水若鸿,陆洪芬,李小秋:病理诊断复核,研究指导;
于宝华:研究选题,设计研究方案,修改文章。
[参考文献]
[1]LIU Z Y, ZHANG Y L, ZHU Y C, et al. Prognosis of intravascular large B cell lymphoma (IVLBCL): analysis of 182 patients from global case series[J]. Cancer Manag Res, 2020, 12: 10531-10540.
[2]WHO Classification of Tumours Editorial Board. Haematolymphoid tumours (5th ed)[M]. Lyon (France): International Agency for Research on Cancer, 2024.
[3]HANS C P, WEISENBURGER D D, GREINER T C, et al. Confirmation of the molecular classification of diffuse large B-cell lymphoma by immunohistochemistry using a tissue microarray[J]. Blood, 2004, 103(1): 275-282.
[4]WAWIRE J, SAYED S, MOLOO Z, et al. Diffuse large B-cell lymphoma in Kenya: MYC, BCL2, and the cell of origin[J]. J Glob Oncol, 2019, 5: 1-8.
[5]CAMPUZANO-ZULUAGA G, CIOFFI-LAVINA M, LOSSOS I S, et al. Frequency and extent of CD30 expression in diffuse large B-cell lymphoma and its relation to clinical and biologic factors: a retrospective study of 167 cases[J]. Leuk Lymphoma, 2013, 54(11): 2405-2411.
[6]SWERDLOW S H, CAMPO E, HARRIS N L, et al. World Health Organization classification of tumors of haematopoietic and lymphoid tissues[M]. Lyon: International Agency for Research on Cancer, 2008.
[7]MURASE T, YAMAGUCHI M, SUZUKI R, et al. Intravascular large B-cell lymphoma (IVLBCL): a clinicopathologic study of 96 cases with special reference to the immunophenotypic heterogeneity of CD5[J]. Blood, 2007, 109(2): 478-485.
[8]中华医学会血液学分会淋巴细胞疾病学组, 中国临床肿瘤学会(CSCO)淋巴瘤专家委员会. 血管内大B细胞淋巴瘤诊治中国专家共识(2023年版)[J].中华血液学杂志, 2023, 44(3): 177-181.
Lymphoid Disease Group, Chinese Society of Hematology, Chinese Medical Association; Lymphoma Expert Committee of Chinese Society of Clinical Oncology (CSCO). Chinese expert consensus on the diagnosis and management of intravascular large B cell lymphoma (2023)[J]. Chin J Hematol, 2023, 44(3): 177-181.
[9]MATSUE K, ABE Y, NARITA K, et al. Diagnosis of intravascular large B cell lymphoma: novel insights into clinicopathological features from 42 patients at a single institution over 20years[J]. Br J Haematol, 2019, 187(3): 328-336.
[10]KIRIAKOPOULOS A, LINOS D. Intravascular B-large cell lymphoma: an unexpected diagnosis of an incidental adrenal mass[J]. J Surg Case Rep, 2019, 2019(2): rjz048.
[11]FUKUSHIMA A, OKADA Y, TANIKAWA T, et al. Primary bilateral adrenal intravascular large B-cell lymphoma associated with adrenal failure[J]. Intern Med, 2003, 42(7): 609-614.
[12]YU Y, GOVINDARAJAN R. Intravascular large B-cell lymphoma presenting as an isolated cauda equina-conus medullaris syndrome-a case report[J]. J Spinal Cord Med, 2020, 43(4): 556-559.
[13]TAKAHASHI Y, IIDA K, HINO Y, et al. Silent intravascular lymphoma initially manifesting as a unilateral adrenal incidentaloma[J]. Case Rep Med, 2012, 2012: 849285.
[14]LI W, LIN W, MA C, et al. A case of intravascular large B-cell lymphoma in the left adrenal and another tumor in the right adrenal detected by (18)F-FDG PET/CT[J]. Hell J Nucl Med, 2016, 19(1): 57-59.
[15]CUI J, LIU Q, CHENG Y X, et al. An intravascular large B-cell lymphoma with a t(3;14)(q27;q32) translocation[J]. J Clin Pathol, 2014, 67(3): 279-281.
[16]SRIVATSA S, SHARMA J, LOGANI S. Intravascular lymphoma: an unusual diagnostic outcome of an incidentally detected adrenal mass[J]. Endocr Pract, 2008, 14(7): 884-888.
[17]VENIZELOS I, TAMIOLAKIS D, PETRAKIS G. High grade primary adrenal intravascular large B-cell lymphoma manifesting as Addison disease[J]. Rev Esp Enferm Dig, 2007, 99(8): 471-474.
[18] ASKARIAN F, XU D S. Adrenal enlargement and insufficiency: a common presentation of intravascular large B-cell lymphoma[J]. Am J Hematol, 2006, 81(6): 411-413.
[19] COLAVOLPE C, EBBO M, TROUSSE D, et al. FDG-PET/CT is a pivotal imaging modality to diagnose rare intravascular large B-cell lymphoma: case report and review of literature[J]. Hematol Oncol, 2015, 33(2): 99-109.
[20] SCHRADER A M R, JANSEN P M, WILLEMZE R, et al. High prevalence of MYD88 and CD79B mutations in intravascular large B-cell lymphoma[J]. Blood, 2018, 131(18): 2086-2089.
[21] BOONSAKAN P, IAMSUMANG W, CHANTRATHAMMACHART P, et al. Prognostic value of concurrent expression of C-MYC and BCL2 in intravascular large B-cell lymphoma: a 10-year retrospective study[J]. Biomed Res Int, 2020, 2020: 1350820.
[22] YEGAPPAN S, COUPLAND R, ARBER D A, et al. Angiotropic lymphoma: an immunophenotypically and clinically heterogeneous lymphoma[J]. Mod Pathol, 2001, 14(11): 1147-1156.
[23] YAMAGUCHI M, SETO M, OKAMOTO M, et al. De novo CD5+ diffuse large B-cell lymphoma: a clinicopathologic study of 109 patients[J]. Blood, 2002, 99(3): 815-821.
[24] GUPTA G K, JAFFE E S, PITTALUGA S. A study of PD-L1 expression in intravascular large B cell lymphoma: correlation with clinical and pathological features[J]. Histopathology, 2019, 75(2): 282-286.
[25] SUEHARA Y, SAKATA-YANAGIMOTO M, HATTORI K, et al. Liquid biopsy for the identification of intravascular large B-cell lymphoma[J]. Haematologica, 2018, 103(6): e241-e244.
[26] SHIMADA K, YOSHIDA K, SUZUKI Y, et al. Frequent genetic alterations in immune checkpoint-related genes in intravascular large B-cell lymphoma[J]. Blood, 2021, 137(11): 1491-1502.
[27] WILSON W H, YOUNG R M, SCHMITZ R, et al. Targeting B cell receptor signaling with ibrutinib in diffuse large B cell lymphoma[J]. Nat Med, 2015, 21(8): 922-926.
[28] XIE J L, SHEN X, SHI Q, et al. Clinical significance of MYD88 non-L265P mutations in diffuse large B-cell lymphoma[J]. Hematol Oncol, 2022, 40(5): 885-893.
[29] JIANG S Y, QIN Y, GUI L, et al. Genomic alterations and MYD88MUT variant mapping in patients with diffuse large B-cell lymphoma and response to ibrutinib[J]. Target Oncol, 2020, 15(2): 221-230.
[30] ZENZ T, KREUZ M, FUGE, et al. TP53 mutation and survival in aggressive B cell lymphoma[J]. Int J Cancer, 2017, 141(7): 1381-1388.
[31] JACOBS G, HELLMIG S, HUSE K, et al. Polymorphisms in the 3’-untranslated region of the CDH1 gene are a risk factor for primary gastric diffuse large B-cell lymphoma[J]. Haematologica, 2011, 96(7): 987-995.
[32] CARRERAS J, IKOMA H, KIKUTI Y Y, et al. Mutational, immune microenvironment, and clinicopathological profiles of diffuse large B-cell lymphoma and follicular lymphoma with BCL6 rearrangement[J]. Virchows Arch, 2024, 484(4): 657-676.
[33] DE MIRANDA N F, PENG R J, GEORGIOU K, et al. DNA repair genes are selectively mutated in diffuse large B cell lymphomas[J]. J Exp Med, 2013, 210(9): 1729-1742.
[34] ZHANG Y, JIA C W, WANG W, et al. The interim analysis from a prospective single-center phase 2 study of zanubrutinib plus R-CHOP in treat-Naïve intravascular large B cell lymphoma[J]. Blood, 2021, 138(Supplement 1): 3563.
[35] RAJYAGURU D J, BHASKAR C, BORGERT A J, et al. Intravascular large B-cell lymphoma in the United States (US): a population-based study using Surveillance, Epidemiology, and End Results program and National Cancer Database[J]. Leuk Lymphoma, 2017, 58(9): 1-9.
- 搜索
-
- 1000℃Nutrients:真实世界数据,纤维肌痛患者的饮食与运动自适应规律
- 1000℃D-二聚体升高诊治与管理专家共识(2026)
- 1000℃专家论坛|文良志:门静脉血栓的诊断和治疗
- 1000℃首例儿童NF2驱动型胸膜间皮瘤,多方法学检测锁定NF2双等位基因失活和14/22号染色体缺失,提示与成人胸膜间皮瘤不同
- 1000℃打破误区:干扰素追求CHB功能性治愈,HBsAg为何“不降反增”?
- 1000℃迷惑性极强的肝内病灶!影像表现疑点重重,最终病理竟查出两种不同肝脏恶性肿瘤
- 1000℃指南共识|原发性肝癌分子靶向药物相关蛋白尿中西医结合诊疗专家共识
- 1000℃Diabetologia:意大利北部社区 1~100 岁人群胰岛自身抗体与乳糜泻 TGA-IgA 的年龄分布及检测方法学验证
- 精J Child Psychol Psychiatry:12种罕见神经发育障碍儿童沟通能力谱系
- 精研究发现:爱吃辣的人,心血管病和癌症死亡风险都会显著降低
- 精Nursing in Critical Care:别再指责护士了!ICU 里被遗漏的护理,根源在系统而非个人
- 精Acta Obstet Gynecol Scand:罕见病女性的妊娠并发症与母婴结局,一项单中心434种罕见病的回顾性队列研究
- 精【爱儿小醉】儿科患者术前对流层臭氧暴露与围手术期呼吸系统不良事件之间的关系:一项单中心回顾性队列研究
- 精eBioMedicine:牙龈下微生物组与脑健康存在连续关联梯度,牙周炎或成认知衰退可干预靶点
- 精军事医学研究院《自然·通讯》:自适应IrPtCu纳米酶水凝胶实现耐药菌感染伤口序贯治疗
- 精能够逆转萎缩性胃炎的两个中成药,该怎么选择?
- 荐同时性多发性原发性肺癌,左右病灶分别为EGFR和ALK阳性,考虑淋巴结肿大仅局限左肺门及血浆EGFR阳性,采用奥希替尼联合化疗
- 荐40岁女性同时罹患卵巢支持细胞-间质细胞瘤和透明细胞乳头状肾肿瘤,WES等基因检测竟为阴性
- 荐椎管内麻醉使用止血药突发气道痉挛的抢救流程解析
- 荐女子肝区无任何不适,影像提示复杂囊性病变,层层鉴别后锁定罕见胆管源性囊性肿瘤
- 荐“绘”真报告 | 病理考虑为中枢神经细胞瘤,检出脑室外神经细胞瘤的特征性变异FGFR1-TACC1融合,辅助鉴别诊断
- 荐8岁女童出现男性化症状,竟是形似「性索-间质肿瘤」的卵巢「无性细胞瘤」所致,少见KRAS/CDK4共扩增或与侵袭性有关
- 荐17例病例分析揭示常见于中年人的色素性室管膜瘤临床特征与预后,分子检测可助力临床精准诊疗
- 荐Lancet Oncol:结直肠癌腹膜转移,围手术期化疗并非必选项
- 标签列表
-
- 星座 (702)
- 孩子 (526)
- 恋爱 (505)
- 婴儿车 (390)
- 宝宝 (328)
- 狮子座 (313)
- 金牛座 (313)
- 摩羯座 (302)
- 白羊座 (301)
- 天蝎座 (294)
- 巨蟹座 (289)
- 双子座 (289)
- 处女座 (285)
- 天秤座 (276)
- 双鱼座 (268)
- 婴儿 (265)
- 水瓶座 (260)
- 射手座 (239)
- 不完美妈妈 (173)
- 跳槽那些事儿 (168)
- baby (140)
- 女婴 (132)
- 生肖 (129)
- 女儿 (129)
- 民警 (127)
- 狮子 (105)
- NBA (101)
- 家长 (97)
- 怀孕 (95)
- 儿童 (93)
- 交警 (89)
- 孕妇 (77)
- 儿子 (75)
- Angelababy (74)
- 父母 (74)
- 幼儿园 (73)
- 医院 (69)
- 童车 (66)
- 女子 (60)
- 郑州 (58)