首页 > 医疗资讯/ 正文
论坛导读:运动神经元病(Motor Neuron Disease, MND)是一组以运动神经元退行性死亡为特征的致死性神经系统疾病,包括肌萎缩侧索硬化(ALS)、脊髓性肌萎缩症(SMA)等亚型。近年来,随着分子生物学、神经电生理学及基因组学等领域的突破,MND的基础研究与临床诊疗均取得显著进展。

运动神经元病(motor neuron disease,MND)通常分为肌缩性侧索硬化症(amyotrophic lateral sclerosis,ALS)、进行性肌萎缩(progressive muscle atrophy,PMA)、进行性延髓麻痹(progressive bulbar palsy,PBP)、原发性侧索硬化(primary lateral sclerosis,PLS)四种类型。其他少见类型如连枷臂综合征(FAS)和连枷腿综合征(FLS),FAS和FLS均表现为症状和体征局限于肢体1个区域达12个月以上而不出现其他区域受累的体征。
MND是一类累及上下运动神经元的慢性进行性神经系统疾病,以肌肉萎缩、无力及运动功能障碍为主要表现,最终因呼吸衰竭导致死亡。其病因复杂,涉及遗传、环境及氧化应激等多因素交互作用。据流行病学统计,MND全球发病率约为2/10万,患病率约4-6/10万,其中ALS占80%以上。尽管目前尚无根治方法,但基础研究的深入与临床技术的创新为延缓疾病进展、改善患者生活质量提供了新希望。
ALS是运动神经元病中最常见的类型,一般中老年发病多见,我国ALS发病年龄高峰在50岁左右,并且发病年龄有年轻化趋势,少数患者可20岁左右即发病。该病呈持续进展,目前尚无有效的治疗方法,护理和呼吸支持对患者的生活质量、生存率具有明显改善。

MND的病理生理机制
1. 遗传学基础
MND具有显著的遗传异质性。已发现超过50个致病基因,包括SOD1、TARDBP、FUS及C9orf72等。其中,C9orf72基因的内含子六核苷酸重复扩增是家族性ALS(fALS)最常见的遗传病因,占欧美人群的40%-50%。这些基因突变通过干扰RNA代谢、蛋白质稳态及自噬功能等途径,导致运动神经元选择性死亡。
2. 分子与细胞机制
兴奋性谷氨酸过度激活NMDA受体,引发钙超载和线粒体损伤,是ALS早期病理特征。 蛋白质错误折叠与聚集,如SOD1、TDP-43等蛋白异常聚集形成病理性包涵体,干扰神经元正常功能。
- 氧化应激与线粒体功能障碍:活性氧(ROS)累积导致脂质、DNA及蛋白质氧化损伤,线粒体膜电位下降加速神经元凋亡。
- 神经炎症反应:小胶质细胞过度活化释放促炎因子(如TNF-α、IL-1β),加剧神经元损伤。
3. 轴突运输障碍
运动神经元的长轴突依赖高效的微管运输系统。突变基因(如DYNC1H1)导致的动力蛋白异常,使神经营养因子(如GDNF)逆向运输受阻,加速远端轴突退变。
MND的诊断技术进展
1. 临床评估与神经电生理检查
肌电图(EMG)与神经传导速度(NCV)中EMG可检测失神经支配(纤颤电位、正锐波),NCV用于区分脱髓鞘与轴索性病变。 运动诱发电位(MEP)评估上运动神经元损伤,MEP缺失提示皮质脊髓束病变。
2. 影像学技术
磁共振成像(MRI)T2加权像显示皮质脊髓束高信号(“虎斑征”),弥散张量成像(DTI)量化白质纤维完整性。 正电子发射断层扫描(PET)通过代谢成像(如18F-FDG)评估脑区功能异常,辅助早期诊断。
3. 生物标志物研究
脑脊液(CSF)标志物TDP-43、NfL(神经丝轻链)水平升高与疾病进展相关。 血液外泌体miRNA,如miR-206、miR-455等作为潜在无创诊断标志物,已进入临床试验验证阶段。
4. 人工智能辅助诊断
基于深度学习的EMG信号分析模型(如CNN-LSTM)可自动识别运动神经元损伤特征,诊断准确率达85%以上。
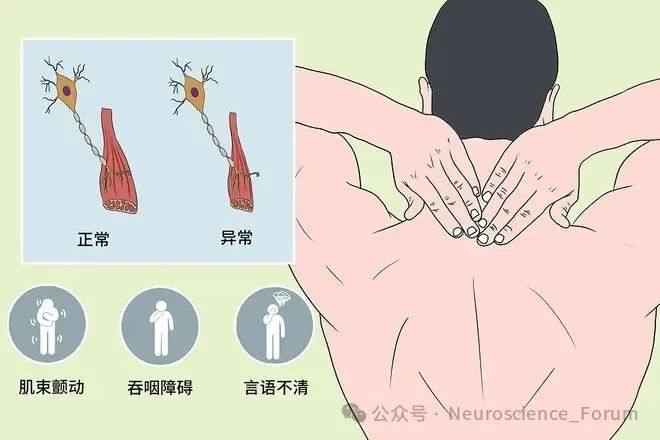
MND的治疗策略与临床实践
1. 药物治疗
谷氨酸受体拮抗剂利鲁唑(Riluzole)通过抑制突触前谷氨酸释放,延长患者生存期3-6个月,是唯一获批的ALS治疗药物。自由基清除剂依达拉奉(Edaravone)通过清除ROS减轻氧化损伤,适用于早期ALS患者。
2. 细胞与基因治疗
诱导多能干细胞(iPSC)分化的运动神经元可修复受损组织,目前处于Ⅰ/Ⅱ期临床试验阶段。反义寡核苷酸(ASO)针对C9orf72基因的ASO药物(如Tofersen)可减少毒性蛋白聚集,已获FDA快速通道资格。
Tofersen注射液是一种反义寡核苷酸(ASO)药物,可通过减少SOD1蛋白合成,减少毒性SOD1蛋白的蓄积,从而减轻运动神经元的损伤,减缓疾病进展。虽然托夫生注射液仅针对发生SOD1基因突变的渐冻症患者,但它的获批也深深鼓舞了整个渐冻症患者群体,让他们重新燃起了生命的希望,也极大提振了整个渐冻症领域的研发信心,为更多创新疗法的诞生奠定了基础。
3. 康复与支持治疗
无创正压通气(NIV)可延长生存期,膈肌起搏器改善呼吸肌无力。经皮内镜下胃造瘘术(PEG)预防误吸性肺炎,维持患者营养状态。
4. 中西医结合治疗
中医将MND归为“痿证”,以“治痿独取阳明”为原则。齐向华教授基于系统辨证脉学,将ALS分为气机郁滞、气虚痰瘀、气机下陷三型,分别采用柴胡疏肝散、瓜蒌薤白半夏汤等方剂,临床显示可改善肌力与生存质量。
挑战与未来研究方向
1. 病理机制未明
约90%的MND为散发性,其环境诱因(如重金属暴露、运动损伤)与遗传易感性的交互机制仍需深入探索。
2. 治疗瓶颈
现有药物仅能延缓病程,无法阻止神经元死亡。未来需开发靶向轴突再生、神经炎症调控及蛋白稳态恢复的多靶点疗法。
3. 精准医疗与个体化治疗
基于基因分型(如C9orf72突变型)的定制化治疗方案将成为主流。AI驱动的生物标志物动态监测平台可实时评估疗效。
4. 跨学科协作与国际合作
建立全球MND数据库(如PRO-ACT),整合临床、基因组及影像数据,加速新药研发与临床试验设计。
结论
MND是一种成人起病的致死性神经系统退行性疾病,可分为肢体起病型ALS、球部起病型ALS、PMA、PLS、FAS、FLS等多种类型,其中绝大部分患者可按照修订版El Escorial诊断标准或Awaji-shima诊断标准进行诊断。目前本病尚无有效逆转疾病的方法,但包括营养支持、呼吸管理及药物治疗在内的综合治疗,可以在一定程度上延缓疾病进程,延长患者的生存时间并提高患者生活质量。目前运动神经元病的基础与临床研究正从单一病理机制探索转向多组学整合与精准干预。尽管挑战重重,但基因编辑、干细胞技术及人工智能的突破为攻克这一顽疾提供了全新视角。未来需加强基础-临床转化,推动全球协作,最终实现MND的可防、可治与可控。
- 搜索
-
- 1000℃Nutrients:真实世界数据,纤维肌痛患者的饮食与运动自适应规律
- 1000℃D-二聚体升高诊治与管理专家共识(2026)
- 1000℃专家论坛|文良志:门静脉血栓的诊断和治疗
- 1000℃首例儿童NF2驱动型胸膜间皮瘤,多方法学检测锁定NF2双等位基因失活和14/22号染色体缺失,提示与成人胸膜间皮瘤不同
- 1000℃打破误区:干扰素追求CHB功能性治愈,HBsAg为何“不降反增”?
- 1000℃迷惑性极强的肝内病灶!影像表现疑点重重,最终病理竟查出两种不同肝脏恶性肿瘤
- 1000℃指南共识|原发性肝癌分子靶向药物相关蛋白尿中西医结合诊疗专家共识
- 1000℃Diabetologia:意大利北部社区 1~100 岁人群胰岛自身抗体与乳糜泻 TGA-IgA 的年龄分布及检测方法学验证
- 精J Child Psychol Psychiatry:12种罕见神经发育障碍儿童沟通能力谱系
- 精研究发现:爱吃辣的人,心血管病和癌症死亡风险都会显著降低
- 精Nursing in Critical Care:别再指责护士了!ICU 里被遗漏的护理,根源在系统而非个人
- 精Acta Obstet Gynecol Scand:罕见病女性的妊娠并发症与母婴结局,一项单中心434种罕见病的回顾性队列研究
- 精【爱儿小醉】儿科患者术前对流层臭氧暴露与围手术期呼吸系统不良事件之间的关系:一项单中心回顾性队列研究
- 精eBioMedicine:牙龈下微生物组与脑健康存在连续关联梯度,牙周炎或成认知衰退可干预靶点
- 精军事医学研究院《自然·通讯》:自适应IrPtCu纳米酶水凝胶实现耐药菌感染伤口序贯治疗
- 精能够逆转萎缩性胃炎的两个中成药,该怎么选择?
- 荐同时性多发性原发性肺癌,左右病灶分别为EGFR和ALK阳性,考虑淋巴结肿大仅局限左肺门及血浆EGFR阳性,采用奥希替尼联合化疗
- 荐40岁女性同时罹患卵巢支持细胞-间质细胞瘤和透明细胞乳头状肾肿瘤,WES等基因检测竟为阴性
- 荐椎管内麻醉使用止血药突发气道痉挛的抢救流程解析
- 荐女子肝区无任何不适,影像提示复杂囊性病变,层层鉴别后锁定罕见胆管源性囊性肿瘤
- 荐“绘”真报告 | 病理考虑为中枢神经细胞瘤,检出脑室外神经细胞瘤的特征性变异FGFR1-TACC1融合,辅助鉴别诊断
- 荐8岁女童出现男性化症状,竟是形似「性索-间质肿瘤」的卵巢「无性细胞瘤」所致,少见KRAS/CDK4共扩增或与侵袭性有关
- 荐17例病例分析揭示常见于中年人的色素性室管膜瘤临床特征与预后,分子检测可助力临床精准诊疗
- 荐Lancet Oncol:结直肠癌腹膜转移,围手术期化疗并非必选项
- 标签列表
-
- 星座 (702)
- 孩子 (526)
- 恋爱 (505)
- 婴儿车 (390)
- 宝宝 (328)
- 狮子座 (313)
- 金牛座 (313)
- 摩羯座 (302)
- 白羊座 (301)
- 天蝎座 (294)
- 巨蟹座 (289)
- 双子座 (289)
- 处女座 (285)
- 天秤座 (276)
- 双鱼座 (268)
- 婴儿 (265)
- 水瓶座 (260)
- 射手座 (239)
- 不完美妈妈 (173)
- 跳槽那些事儿 (168)
- baby (140)
- 女婴 (132)
- 生肖 (129)
- 女儿 (129)
- 民警 (127)
- 狮子 (105)
- NBA (101)
- 家长 (97)
- 怀孕 (95)
- 儿童 (93)
- 交警 (89)
- 孕妇 (77)
- 儿子 (75)
- Angelababy (74)
- 父母 (74)
- 幼儿园 (73)
- 医院 (69)
- 童车 (66)
- 女子 (60)
- 郑州 (58)