首页 > 医疗资讯/ 正文
摘 要
目的 对3个遗传性痉挛性截瘫(hereditary spastic paraplegia,HSP)患者家系的临床表现及致病基因进行分析。
方法 对甘肃省妇幼保健院(甘肃省中心医院)收集的3个HSP患者家系进行基因分析。
结果 家系1中先证者为FA2H c.159_176delGGCGGGCCAGGACATCAG (p.Arg53_Ser59delinsSer)纯合变异导致的常染色体隐性痉挛性截瘫35型,家系2中的先证者为AP4B1 c.1399G>T(p.Glu467Ter)纯合变异导致的常染色体隐性痉挛性截瘫47型,家系3中先证者为SPG11 c.7023C>G(p.Tyr2341Ter)的纯合变异导致的常染色体隐性痉挛性截瘫11型。其中,AP4B1 c.1399G>T(p.Glu467Ter)位点为尚未报告的变异。根据美国医学遗传学与基因组学学会(American College of Medical Genetics and Genomics,ACMG)指南,该变异的致病性评级为致病性变异。
结论 本研究丰富了HSP致病基因AP4B1的变异谱,为提高临床对HSP患者的认识与诊断能力提供了基础性数据。
关键词
遗传性痉挛性截瘫;基因变异;基因诊断;干预治疗;临床诊断;常染色体隐性;FA2H基因;AP4B1基因;SPG11基因
遗传性痉挛性截瘫(hereditary spastic paraplegia,HSP)是一类罕见的神经退行性疾病[1]。HSP患者可能表现出广泛的症状,从非常轻微的下肢痉挛到严重的神经和非神经表现。这些症状经常与其他疾病重叠,这可能导致错误的诊断[2-3]。因此,需要进行基因检测来进行分型诊断,与多种具有表型和基因型的重叠遗传性疾病进行鉴别诊断。目前,已明确的致病基因超过90个,遗传方式包括常染色体显性遗传(autosomal dominant,AD)、常染色体隐性遗传(autosomal recessive,AR)、X染色体连锁遗传(X-linked inheritance,XL)以及线粒体遗传[4]。本研究通过对3个HSP患者家系的临床表现及致病基因进行分析,以提高临床对HSP患者的认识与诊断能力。
1 对象与方法
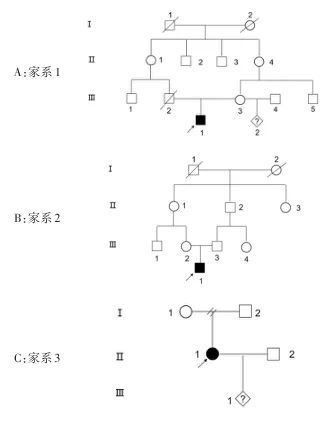
1.1 研究对象 家系1:患儿,男10岁,足月顺产,行走不稳4年余。0~5岁未见显著异常;5岁一次意外(摔伤),半年后出现行为特殊,运动能力全面下降,逐渐出现行走不稳,且进行性加重,表现为行走不稳,呈慌张步态,脚后跟不着地,走路严重不协调,双下肢及张力增高明显,同时伴有右手握力弱等症状。爱笑,智力尚可,精神状态差,久坐后易瘫倒。反应慢,晚上睡觉蜷缩状;外院MRI提示:未见明显异常(未见报告),建议先证者行头颅MRI检查,家属拒绝,父母为表兄妹(图1A),因母亲再婚拟生育二胎前来就诊。
家系2:先证者,男,18岁,足月顺产,1岁发现脑瘫。2岁不会站,不会走,不会说话;脑电图提示不正常脑电图;外院头颅MRI平扫提示胼胝体发育不良,右侧乳突炎,脑室扩大,交通性脑积水。父母为表兄妹(图1B),因父母拟生育二胎前来就诊。
家系3:患者,女,24岁,15岁时发现行走异常,渐渐加重,外院脑部MRI检查提示:脑桥后不对称性点状稍长T2信号;双侧额颞顶部脑外间隙增宽;双侧海马体积稍缩小;胼胝体发育不良。行康复治疗,尚可缓慢行走,孕期行动较困难,可扶行,说话吐字不清。父母离异(图1C),患者因怀孕前来进行基因诊断,明确胎儿患病风险。
本研究通过了甘肃省妇幼保健院伦理委员会审查[(2021)GSFY伦审65号],家系成员均签署了临床研究知情同意书。
图1 患者家系图Fig.1 Pedigree of the patients
1.2 方法
1.2.1 基因组DNA提取 采集患者及其父母外周静脉血2~3 mL,EDTA抗凝。应用天根生化科技(北京)有限公司生产的血液基因组DNA提取试剂盒(货号:DP348)提取基因组DNA。
1.2.2 高通量测序分析 采用目标区域捕获-WES检测技术进行检测,目标区域 20×以上覆盖度达到 95%。检测区域包括外显子区和剪接区、部分内含子区;可检测突变类型包括非重复区域的单碱基突变(single-nucleotide variant,SNV)、小片段插入/缺失(≤20 bp)(insertion/deletion,INDEL)以及部分外显子缺失重复和CNV检测。
1.2.3 数据分析 高通量测序得到的结果与参考基因组(GRCp7/hg19)比对得到所有变异的 SNP_INDEL文件,对检测到的碱基改变进行筛选,应用PolyPhen2(http://genetics.bwh.harvard.edu/ppp/)和PROVEAN(http://provean.jcvi.org/index.php) 软件对新变异进行蛋白功能预测,最后依据美国医学遗传学与基因组学学会(American College of Medical genetics and Genomics,ACMG)指南[5]判读新变异的致病性。
1.2.4 Sanger测序验证 可疑基因致病变异采用Sanger测序验证,基因外显子扩增引物由生工生物工程(上海)股份有限公司合成。PCR扩增体系为25 μL:其中2×Taq PCR Mastermix[天根生化科技(北京)有限公司]12.5 μL,基因组DNA模板1.5 μL(浓度20~50 ng/μL),上下游引物(10 μmol/L)各 0.5 μL,ddH2O 10 μL。 PCR扩增产物经琼脂电泳鉴定后对目的条带单一的产物采用PCR产物纯化试剂盒[天根生化科技(北京)有限公司]纯化后,由ABI 3500型(ABI,美国)测序仪进行测序,使用SeqMan软件与标准序列对测序结果进行比对分析。
2 结果
2.1 基因分析结果 家系1:先证者检测到FA2H c.159_176delGGCGGGCCAGGACATCAG (p.Arg53_Ser59delinsSer)纯合变异,关联疾病为常染色体隐性痉挛性截瘫35型(OMIM:612319)。根据ACMG变异评级指南,该变异评级为疑似致病变异,致病证据为:PS3+PM2+PM4(表1),即已有文献报告体内、体外功能实验已明确会导致基因功能受损(PS3)[6],所有正常人群数据库频率小于0.0005(PM2),非重复区框内插入/缺失或终止密码子丧失导致的蛋白质长度变化(PM4)。
表1 患者临床及疾病诊断信息表Tab.1 Clinical and disease diagnosis information of the patients
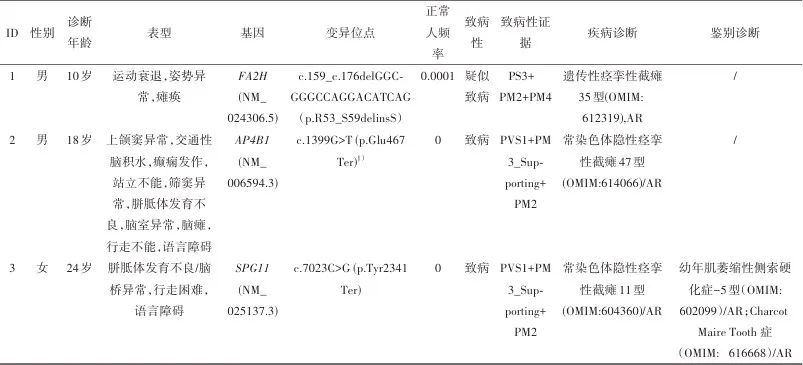
注:1)未报道的新变异。
家系2:先证者检测到AP4B1 c.1399G>T(p.Glu467Ter)纯合变异,为未报告过的新变异,关联疾病为常染色体隐性痉挛性截瘫47型(OMIM:614066)。根据ACMG变异评级指南,该变异评级为致病变异,致病证据为:PVS1+PM3_Supporting+PM2(表1),即该变异为无义变异(PVS1),在隐性遗传病中,在反式位置上检测到致病变异(PM3_Supporting),所有正常人群数据库频率小于0.0005(PM2)。
家系3:先证者检测到SPG11 c.7023C>G(p.Tyr2341Ter)的纯合变异,关联疾病为常染色体隐性痉挛性截瘫11型(OMIM:604360)。根据ACMG变异评级指南,该变异评级为致病变异,致病证据为:PVS1+PM3_Supporting+PM2(表1),即该变异为无义变异(PVS1),在隐性遗传病中,在反式位置上检测到致病变异(PM3_Supporting),所有正常人群数据库频率小于0.0005(PM2)。
2.2 遗传咨询 家系1中先证者诊断为FA2H纯合变异导致常染色体隐性痉挛性截瘫35型,母亲为杂合变异携带者,建议母亲新配偶进行FA2H致病变异位点的筛查,母亲新配偶经单人全外显子组测序,未发现FA2H疑似致病及致病性变异,告知患者母亲,可正常妊娠,但由于是高龄,孕中期需做产前诊断来避免非整倍体的发生。
家系2中先证者诊断为AP4B1 c.1399G>T纯合变异导致的常染色体隐性痉挛性截瘫47型,父母均为杂合变异携带者,告知夫妇可通过自然受孕后孕中期进行产前诊断或者通过三代试管助孕阻断该疾病后,该夫妇决定采用三代试管助孕。
家系3中先证者诊断为SPG11纯合变异导致的常染色体隐性痉挛性截瘫11型,先证者为孕妇,建议对其配偶进行SPG11的致病性变异位点的筛查,其配偶经单人全外显子组测序,未发现SPG11疑似致病及致病性变异。告知患者家属,孕妇的孩子50%的概率是SPG11正常的,50%的概率是SPG11 c.7023C>G(p.Tyr2341Ter)位点的杂合携带者,患病的风险小,可进行常规的产前检查即可。
3 讨论
3.1 HSP的临床特点与临床诊断 HSP患病率约为0.1/10万~9.6/10万,起病多隐匿,以进行性加重的双下肢痉挛无力、步态异常为核心临床特点[7]。HSP可起病于婴儿期、儿童期、青春期和成年期等不同年龄段,根据发病年龄,可分为早发型(<35 岁,Ⅰ型)和晚发型(≥35岁,Ⅱ型)。根据临床特点,HSP可分为“单纯型”和“复杂型”两大类,“单纯型”只有运动功能障碍,以双下肢进行性痉挛无力、僵硬、步态异常为核心特点,或可能伴随尿便障碍、深感觉障碍等;“复杂型”除了具有上述特点,还可叠加包括智力发育迟滞、构音障碍、视听觉障碍、鱼鳞病、吞咽障碍、癫痫发作、共济失调、周围神经病变、胼胝体异常、脑白质病变以及小脑萎缩等异常症状[8-9]。
HSP的临床诊断主要根据患者是否存在进行性双下肢痉挛无力的临床表现,包括双下肢腱反射亢进、踝阵挛和髌阵挛阳性、Babinski征阳性,以及起病年龄、起病方式、既往病史、家族史等[4]。此外,需与继发因素所引起的慢性脊髓病变进行鉴别诊断,如脊髓外伤、脊髓空洞症、脊髓压迫症、多发性硬化、中枢神经系统感染、微量元素缺乏等。还应与其他与HSP有类似临床表现的脑白质营养不良、脊髓小脑性共济失调、肌萎缩侧索硬化症、及生物素酶缺乏症等遗传病进行鉴别[10]。
3.2 基因诊断可为HSP明确分型 HSP发病机制复杂、遗传方式多样,致病基因与临床表型高度异质,根据患者表型,进行HSP和其他与痉挛相关遗传疾病的鉴别诊断十分困难,基因检测有助于疾病的诊断。本研究中,家系1中的先证者为FA2H c.159_176delGGCGGGCCAGGACATCAG(p.Arg53_Ser59delinsSer)纯合变异所指的常染色体隐性痉挛性截瘫35型,该位点在一个巴基斯坦家庭患者家系中被报告过[11],该患者家庭先证者最初症状明显出现在3~4岁,后来在5~6岁时患者逐渐不能行走。虽然给患者补充维生素和定期按摩橄榄油来缓解肌肉无力,但并没有缓解症状。这与本研究中患者5岁之前未见明显异常,5岁发病以后逐渐发展为不能行走的临床表型类似(表1)。该基因的致病性变异会损害髓鞘形成,从而导致痉挛性截瘫,本研究结果也为FA2H c.159_176delGGCGGGCCAGGACATCAG(p.Arg53_Ser59delinsSer)位点在痉挛性截瘫病因学中的致病作用提供了额外的证据。
本研究家系2中的先证者为AP4B1 c.1399G>T(p.Glu467Ter)纯合变异导致的常染色体隐性痉挛性截瘫47型,根据ACMG变异评级指南,该变异评级为致病变异(PVS1+PM3_Supporting+PM2)。该位点为未报道过的新变异,截至2025年3月,ClinVar(https://www.ncbi.nlm.nih.gov/clinvar/)数据库收录的AP4B1可能致病/致病性变异位点为58个,其中移码变异28个,无义变异22个,剪接变异6个,错义变异2个。AP4B1有adaptin_N与B2-adapt-app_C两个功能域,已发现的致病性变异位点均集中于adaptin_N结构域,本研究发现c.1399G>T(p.Glu467Ter)位点也位于这个结构域(图2),提示该位点可能会对AP4B1有重要影响。本研究扩大了AP4B1致病变异谱,为今后常染色体隐性痉挛性截瘫47型患者的基因诊断提供了一定的参考。
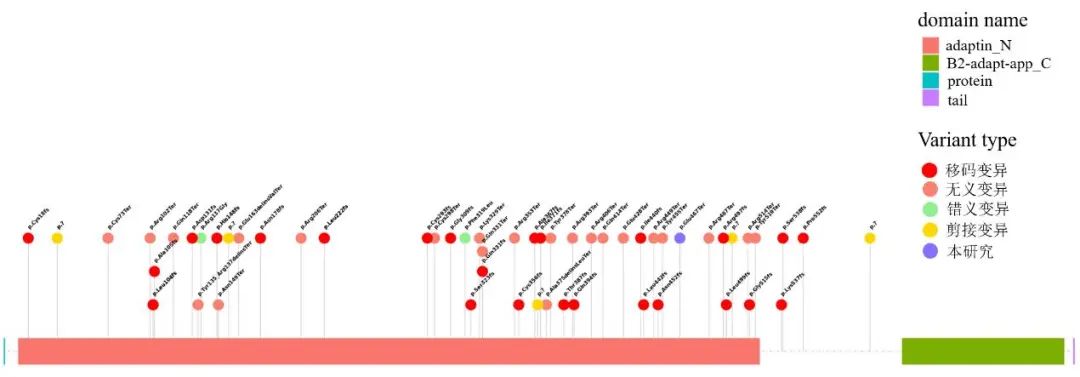
图2 ClinVar数据库已收录AP4B1致病变异位点分布 紫色为本研究发现的c.1399G>T(p.Glu467Ter)。Fig.2 Distribution of pathogenic variants of AP4B1 recorded in the ClinVar database.
本研究中,家系3的先证者为SPG11 c.7023C>G(p.Tyr2341Ter)纯合变异导致的常染色体隐性痉挛性截瘫11型。SPG11导致的痉挛性截瘫11型是最常见的HSP[12],葡萄牙与突尼斯斯法克斯地区该基因导致的HSP占比分别达到了15%与21%[12-13],在中国,SPG11导致的痉挛性截瘫11型也是最常见的[14],其中薄胼胝体是最常见亚型[15]。患者常常有儿童期轻度智力障碍伴学习困难和进行性认知减退、周围神经病变、上肢反射增加、假性延髓受累等症状,少数患者伴有小脑症状、视网膜变性、弓形足、脊柱侧凸、帕金森症等。患者主要在婴儿期或青少年期(1~31岁)发病,多数患者在发病10年或20年后丧失独立行走能力,需依赖于轮椅。本研究中的患者表型与以往报道的患者表型相似,经行康复治疗,24岁尚可缓慢行走,可扶行,说话吐字不清,说明康复治疗科改善患者症状。
3.3 基因诊断可有利于HSP的干预与治疗 目前尚无针对HSP病因有效的治疗方法,多以多学科联合的康复治疗为主,主要通过缓解痉挛及减轻痉挛相关的不适、改善平衡、改善关节活动度、预防和改善异常姿势、延缓肌力下降、提高社会参与能力等来改善生活质量。
基因检测有助于对疑似有HSP患者家族史家庭明确诊断,通过产前诊断或者辅助生殖技术可以预防HSP患者的出生,本研究中的家系2下次妊娠时计划采取辅助生殖助孕来避免患儿的出生,家系1中母亲再婚时配偶与母亲未携带相同致病基因,家系3中孕妇患者的配也未携带与孕妇相同的致病基因,这两个家庭生育与先证者一样的HSP的概率小,可以常规受孕与产检。本研究家系1与家系2的父母均为近亲婚配,由于近亲婚配可能会增加后代遗传病的风险,对于近亲婚配的家庭,一定要做好遗传咨询,可以通过携带者筛查来明确是否为遗传病的高风险夫妇,如果携带同一种遗传病的致病基因或者女方为X连锁隐性遗传病的携带者,一定要做好生育指导,可通过产前诊断或者辅助生殖来避免遗传病患儿的出生。
对已确诊的患者进行全面评估有利于疾病的治疗,随着基因治疗的发展,对HSP各亚型发病机制研究的不断深入,将有希望为HSP患者提供精准的个性化康复治疗方案[16-17]。
3.4 局限性 AP4B1编码适配器相关蛋白复合体4的β1亚基,这个复合体参与细胞内蛋白质的运输和分选。AP4B1 c.1399G>T(p.Glu467Ter)为无义变异,无义变异致病的原理主要涉及基因突变导致蛋白质合成提前终止,进而引发功能异常或缺失。由于AP4B1主要在肾上腺、心脏、肾脏、肺及胃部组织中表达,在外周血中几乎不表达,本研究尚未进行相关功能研究,今后尚需进一步通过体外实验明确该变异位点的潜在致病机制。
综上,本研究发现了一个AP4B1未被报告过的新变异,扩大了AP4B1致病变异谱。本研究通过对3个HSP患者家系的临床表现及致病基因进行分析,为这3个家庭进行了疾病精准诊断与遗传咨询,为HSP的分子遗传学诊断、干预治疗、生育指导及遗传咨询提供了理论依据与技术支持。
参考文献:
1. ESTIAR M A, YU E, HAJ S I, et al. Evidence for Non-Mendelian Inheritance in Spastic Paraplegia 7[J]. Mov Disord, 2021, 36(7): 1664-1675.
2. DIOMEDI M, GAN-OR Z, PLACIDI F, et al. A 23 years follow-up study identifies GLUT1 deficiency syndrome initially diagnosed as complicated hereditary spastic paraplegia[J]. Eur J Med Genet, 2016, 59(11): 564-568.
3. LEVEILLE E, GONORAZKY H D, RIOUX M F, et al. Triple A syndrome presenting as complicated hereditary spastic paraplegia[J]. Mol Genet Genomic Med, 2018, 6(6): 1134-1139.
4. 姚莉,田沃土,曹立.遗传性痉挛性截瘫诊断策略[J].中国神经精神疾病杂志,2023,49(2): 112-119.
5. RICHARDS S, AZIZ N, BALE S, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology[J]. Genet Med, 2015, 17(5): 405-424.
6. DICK K J, ECKHARDT M, PAISAN-RUIZ C, et al. Mutation of FA2H underlies a complicated form of hereditary spastic paraplegia (SPG35)[J]. Hum Mutat, 2010, 31(4): E1251-1260.
7. FINK J K. Hereditary spastic paraplegia: clinico-pathologic features and emerging molecular mechanisms[J]. Acta Neuropathol, 2013, 126(3): 307-328.
8. HARDING A E. Classification of the hereditary ataxias and paraplegias[J]. Lancet, 1983, 1(8334): 1151-1155.
9. BOUTRY M, MORAIS S, STEVANIN G. Update on the Genetics of Spastic Paraplegias[J]. Curr Neurol Neurosci Rep, 2019, 19(4): 18.
10. SHRIBMAN S, REID E, CROSBY A H, et al. Hereditary spastic paraplegia: from diagnosis to emerging therapeutic approaches[J]. Lancet Neurol, 2019, 18(12): 1136-1146.
11. ABBAS S, BRUGGER B, ZUBAIR M, et al. Exome sequencing of a Pakistani family with spastic paraplegia identified an 18 bp deletion in the cytochrome B5 domain of FA2H[J]. Neurol Res, 2021, 43(2): 133-140.
12. RUANO L, MELO C, SILVA MC, et al. The global epidemiology of hereditary ataxia and spastic paraplegia: a systematic review of prevalence studies[J]. Neuroepidemiology, 2014, 42(3): 174-183.
13. COUTINHO P, RUANO L, LOUREIRO J L, et al. Hereditary ataxia and spastic paraplegia in Portugal: a population-based prevalence study[J]. JAMA Neurol, 2013, 70(6): 746-755.
14. DONG E L, WANG C, WU S, et al. Clinical spectrum and genetic landscape for hereditary spastic paraplegias in China[J]. Mol Neurodegener, 2018, 13(1): 36.
15. KARA E, TUCCI A, MANZONI C, et al. Genetic and phenotypic characterization of complex hereditary spastic paraplegia[J]. Brain, 2016, 139(Pt 7): 1904-1918.
16. 姜红芳, 余永林, 阮雯聪, 等.儿童遗传性痉挛性截瘫临床与康复进展[J].中国实用儿科杂志, 2023, 38(1): 19-23.
17. 张钏, 惠玲, 周秉博, 等.甘肃地区遗传代谢病疾病谱及致病基因分析[J].中国当代儿科杂志, 2024, 26(1): 67-71.
猜你喜欢
- 我国首个脑肿瘤注册登记研究平台正式上线
- 四平市第四人民医院双能X射线骨密度骨龄测定仪中标公告
- 血清铁蛋白(SF)升高原因剖析
- 关节炎患者去医院看病应该挂哪个科
- 神外新锐专辑 | 专访北大一院伊志强:率先开展甲状腺相关视神经病变手术 哪些患者能获益
- 如何养成科学的降压饮食习惯?
- Nature | 基于60万人WGS/WES关联分析,鉴定与克隆性造血表型相关的常见和罕见基因变异
- 为养老“资金池”引源头活水
- Nature Commun:四川大学陈崇等人研究揭示EZH2-TFAP2C轴调控胃鳞状细胞癌发病机制
- J Allergy Clin Immunol Pract:花生贴片VP250显著提升幼儿过敏耐受,安全性佳且局部反应递减
- 搜索
-
- 1000℃Nutrients:真实世界数据,纤维肌痛患者的饮食与运动自适应规律
- 1000℃D-二聚体升高诊治与管理专家共识(2026)
- 1000℃专家论坛|文良志:门静脉血栓的诊断和治疗
- 1000℃首例儿童NF2驱动型胸膜间皮瘤,多方法学检测锁定NF2双等位基因失活和14/22号染色体缺失,提示与成人胸膜间皮瘤不同
- 1000℃打破误区:干扰素追求CHB功能性治愈,HBsAg为何“不降反增”?
- 1000℃迷惑性极强的肝内病灶!影像表现疑点重重,最终病理竟查出两种不同肝脏恶性肿瘤
- 1000℃指南共识|原发性肝癌分子靶向药物相关蛋白尿中西医结合诊疗专家共识
- 1000℃Diabetologia:意大利北部社区 1~100 岁人群胰岛自身抗体与乳糜泻 TGA-IgA 的年龄分布及检测方法学验证
- 精J Child Psychol Psychiatry:12种罕见神经发育障碍儿童沟通能力谱系
- 精研究发现:爱吃辣的人,心血管病和癌症死亡风险都会显著降低
- 精Nursing in Critical Care:别再指责护士了!ICU 里被遗漏的护理,根源在系统而非个人
- 精Acta Obstet Gynecol Scand:罕见病女性的妊娠并发症与母婴结局,一项单中心434种罕见病的回顾性队列研究
- 精【爱儿小醉】儿科患者术前对流层臭氧暴露与围手术期呼吸系统不良事件之间的关系:一项单中心回顾性队列研究
- 精eBioMedicine:牙龈下微生物组与脑健康存在连续关联梯度,牙周炎或成认知衰退可干预靶点
- 精军事医学研究院《自然·通讯》:自适应IrPtCu纳米酶水凝胶实现耐药菌感染伤口序贯治疗
- 精能够逆转萎缩性胃炎的两个中成药,该怎么选择?
- 荐同时性多发性原发性肺癌,左右病灶分别为EGFR和ALK阳性,考虑淋巴结肿大仅局限左肺门及血浆EGFR阳性,采用奥希替尼联合化疗
- 荐40岁女性同时罹患卵巢支持细胞-间质细胞瘤和透明细胞乳头状肾肿瘤,WES等基因检测竟为阴性
- 荐椎管内麻醉使用止血药突发气道痉挛的抢救流程解析
- 荐女子肝区无任何不适,影像提示复杂囊性病变,层层鉴别后锁定罕见胆管源性囊性肿瘤
- 荐“绘”真报告 | 病理考虑为中枢神经细胞瘤,检出脑室外神经细胞瘤的特征性变异FGFR1-TACC1融合,辅助鉴别诊断
- 荐8岁女童出现男性化症状,竟是形似「性索-间质肿瘤」的卵巢「无性细胞瘤」所致,少见KRAS/CDK4共扩增或与侵袭性有关
- 荐17例病例分析揭示常见于中年人的色素性室管膜瘤临床特征与预后,分子检测可助力临床精准诊疗
- 荐Lancet Oncol:结直肠癌腹膜转移,围手术期化疗并非必选项
- 标签列表
-
- 星座 (702)
- 孩子 (526)
- 恋爱 (505)
- 婴儿车 (390)
- 宝宝 (328)
- 狮子座 (313)
- 金牛座 (313)
- 摩羯座 (302)
- 白羊座 (301)
- 天蝎座 (294)
- 巨蟹座 (289)
- 双子座 (289)
- 处女座 (285)
- 天秤座 (276)
- 双鱼座 (268)
- 婴儿 (265)
- 水瓶座 (260)
- 射手座 (239)
- 不完美妈妈 (173)
- 跳槽那些事儿 (168)
- baby (140)
- 女婴 (132)
- 生肖 (129)
- 女儿 (129)
- 民警 (127)
- 狮子 (105)
- NBA (101)
- 家长 (97)
- 怀孕 (95)
- 儿童 (93)
- 交警 (89)
- 孕妇 (77)
- 儿子 (75)
- Angelababy (74)
- 父母 (74)
- 幼儿园 (73)
- 医院 (69)
- 童车 (66)
- 女子 (60)
- 郑州 (58)