首页 > 医疗资讯/ 正文
[摘要] 背景与目的:尿激酶型纤溶酶原激活物受体(urokinase-type plasminogen activator receptor,uPAR)基因扩增与胰腺癌患者的不良预后密切相关。uPAR通过丝裂原活化蛋白激酶(mitogen-activated protein kinases,MAPK)信号转导通路调控胰腺癌细胞的上皮-间质转化(epithelial-mesenchymal transformation,EMT)和化疗抵抗,但具体机制尚未完全阐明。本研究旨在探讨uPAR通过抑制细胞自噬促进胰腺癌细胞增殖、侵袭和化疗抵抗的机制。方法:收集2021年12月—2022年6月在南华大学附属长沙中心医院(长沙市中心医院)接受手术切除和穿刺活检患者的胰腺癌组织标本[获得长沙市中心医院医院伦理委员会批准,编号:2021-S0182,2022-S0084],体外培养胰腺癌患者来源类器官(patient-derived organoids,PDO);使用6种胰腺癌细胞系(AsPC-1、PANC-1、CAPAN-1、CAPAN-2、MIA PaCa-2和PaTu8988T),利用成簇规律间隔短回文重复序列及其关联核酸酶9(clustered regularly interspaced short palindromic repeats/CRISPR-associated nuclease 9,CRISPR/Cas9)技术构建uPAR缺陷模型。采用共聚焦显微镜、蛋白质印迹法(Western blot)、酶联免疫吸附实验(enzyme-linked immunosorbent assay,ELISA)和MTS实验测定细胞增殖、侵袭能力,并分析MAPK和自噬信号通路以及吉西他滨诱导的细胞死亡变化。采用RNA干扰(siRNA)或自噬抑制剂评估联合治疗的协同作用。结果:在AsPC-1细胞中,uPAR敲除后,MTS实验和损伤修复实验结果显示,与野生型细胞相比,克隆细胞的增殖和迁移能力显著降低,对吉西他滨的敏感性降低。uPAR重新表达后,克隆细胞的增殖和侵袭能力恢复,且对吉西他滨的敏感性部分恢复。共聚焦显微镜结果显示,克隆细胞的F-肌动蛋白减少,细胞变得圆滑。Western blot结果显示,与野生型细胞相比,克隆细胞中E-钙黏蛋白(E-cadherin)和Slug表达升高,波形蛋白(vimentin)表达降低;磷酸化-黏着斑激酶(phospho-focal adhesion kinase, p-FAK)、p-p38MAPK和微管相关蛋白1的轻链3B(microtubule-associated protein light chain 3B,LC3B)表达升高(P<0.05)。siRNA结果表明,沉默FAK或p38MAPK或联合自噬抑制均能使克隆细胞对吉西他滨重新敏感,沉默p38MAPK能降低LC3B表达。类器官研究显示,8例类器官对吉西他滨的响应存在差异,uPAR在8例胰腺癌类器官中均有表达,uPAR表达水平与吉西他滨的半数抑制浓度(half-maximal inhibitory concentration,IC50)呈负相关(r2=0.66,P<0.05),3例类器官对吉西他滨联合自噬抑制剂响应良好(P<0.05)。结论:uPAR通过抑制胰腺癌中依赖p38MAPK信号通路的细胞活性,防止FAK介导的耐药性和细胞休眠。本研究提示,uPAR高表达的胰腺癌患者对吉西他滨反应更好,而uPAR低表达且p38MAPK高表达的肿瘤可能从联合使用自噬抑制剂和细胞毒性化疗药物治疗中受益。
[关键词] 胰腺癌;类器官培养;uPAR;自噬;吉西他滨;化疗抵抗
[Abstract] Background and purpose: Amplification of the urokinase plasminogen activator receptor (uPAR) gene is closely associated with poor prognosis in pancreatic cancer patients. uPAR regulates epithelial-mesenchymal transition (EMT) and chemoresistance in pancreatic cancer cells through the mitogen-activated protein kinases (MAPK) signaling pathway, though the specific mechanisms remain unclear. This study aimed to investigate the mechanism by which uPAR promotes proliferation, invasion, and chemoresistance of pancreatic cancer cells by inhibiting autophagy. Methods: Pancreatic cancer tissue samples were collected from patients who underwent surgical rep and biopsy at the Changsha Central Hospital, Affiliated to University of South China (Changsha Central Hospital), between December 2021 and Jun 2022. The study was approved by the Ethics Committee of Changsha Central Hospital (Approval No.: 2021-S0182, 2022-S0084). Patient-derived organoids (PDOs) from pancreatic cancer samples were cultured in vitro. Six pancreatic cancer cell lines (AsPC-1, PANC-1, CAPAN-1, CAPAN-2, MIA PaCa-2 and PaTu8988T) were used in this study. uPAR-deficient models were constructed using clustered regularly interspaced short palindromic repeats (CRISPR) Cas9 technology. Cell proliferation and invasion abilities were measured using confocal microscopy, Western blot, enzyme-linked immunosorbent assay (ELISA), and MTS assays. Changes in MAPK and autophagy signaling pathways and gemcitabine-induced cell death were analyzed. The synergistic effects of combined treatments were evaluated using gene silencing (siRNA) or autophagy inhibitors. Results: In AsPC-1 cells, uPAR knockout significantly reduced the proliferation and migration abilities of clone cells compared to wild-type cells, as shown by MTS assays and wound healing experiments, and decreased sensitivity to gemcitabine (P<0.05). Re-expression of uPAR restored the proliferation and invasion abilities of clone cells and partially restored sensitivity to gemcitabine (P<0.05). Confocal microscopy revealed reduced F-actin and a rounded morphology in clone cells. Western blot analysis showed increased expressions of E-cadherin and Slug, decreased expression of vimentin, and increased expressions of phospho-focal adhesion kinase (p-FAK), p-p38MAPK, and the microtubule-associated protein light chain 3B (LC3B) in clone cells compared to wild-type cells. siRNA results indicated that silencing FAK or p38MAPK or combining autophagy inhibition could resensitize clone cells to gemcitabine (P<0.05), with p38MAPK silencing reducing LC3B expression. Organoid studies showed varying responses to gemcitabine among 8 organoids, all expressing uPAR. uPAR expression levels were negatively correlated with gemcitabine IC50 (r2=0.66, P<0.05). Three organoids responded well to the combination of gemcitabine and autophagy inhibitors (P<0.05). Conclusion: uPAR promotes pancreatic cancer cell activity through the p38MAPK signaling pathway, preventing FAK-mediated resistance and cell dormancy. The study suggests that pancreatic cancer patients with high uPAR expression respond better to gemcitabine, while tumors with low uPAR and high p38MAPK expressions may benefit from combined treatment with autophagy inhibitors and cytotoxic chemotherapy.
[Keywords] Pancreatic cancer; Organoid; uPAR; Autophagy; Gemcitabine; Chemoresistance
胰腺导管腺癌(pancreatic ductal adenocarcinoma,PDAC)是一种预后极差的恶性肿瘤[1]。根据转录特征,胰腺癌可以分为经典型、准间充质型和外分泌型三大亚型[2]。然而,胰腺癌细胞具备高度的可塑性,这主要受表观遗传调控因子如zeste基因增强子同源物2 (enhancer of zeste homolog 2,EZH2)和GATA结合蛋白 6 (GATA-binding protein 6 ,GATA6)的调控[3-4]。这种可塑性增加了治疗的复杂性,特别是在选择靶向治疗方案时[5]。因此,治疗前快速准确地确定PDAC的组织学和分子分类对于制定最佳治疗策略至关重要[6]。
尿激酶型纤溶酶原激活物受体(urokinase-type plasminogen activator receptor,uPAR)水平升高与胰腺癌的早期侵袭、转移以及不良预后密切相关[7-8]。研究[9]表明,uPAR在多种实体肿瘤和血液肿瘤中均表现出类似作用。uPAR是一种缺乏跨膜结构的糖基磷脂酰肌醇(glycosylphosphatidylinositol,GPI)锚定膜受体,主要功能是与其配体uPA结合,促进细胞外基质蛋白的降解,并参与细胞迁移、上皮-间质转化(epithelial-mesenchymal transformation,EMT)、增殖和信号转导[10]。通过与整合素的相互作用,uPAR稳定结合玻连蛋白,激活细胞内的Ras-MAPK通路、黏着斑激酶(Focal adhesion kinase, FAK)和Rho家族小GTP酶Rac[11]。uPAR还能够独立于uPA发挥作用,特别是在机械力传递过程中[12]。
FAK是一种广泛表达的酪氨酸激酶,位于黏附复合物中,传递黏附和生长因子依赖性信号[13]。在肿瘤中,FAK是细胞迁移和增殖的正调节因子。然而,在某些情况下,例如在具有强生长因子信号,如表皮生长因子受体(epidermal growth factor receptor,EGFR)或RAS突变以及持续高水平细胞外信号相关激酶(extracellular regulated protein kinase,ERK)的肿瘤中,FAK作为负调节因子限制细胞迁移,这一过程通过ERK依赖性去磷酸化特定FAK酪氨酸残基实现[14-15]。体外实验显示,FAK的激活可导致胰腺癌细胞对吉西他滨产生内源性耐药性[16]。此外,uPAR的下调或阻断会激活SRC、FAK和p38MAPK,导致细胞周期停滞和休眠[17-18]。
我们的前期研究显示,高水平uPAR与PDAC患者较短的生存期相关,特别是在HNF1A阳性的外分泌样肿瘤亚组中。通过成簇规律间隔的短回文重复序列及其关联核酸酶9(clustered regularly interspaced short palindromic repeats/CRISPR-associated nuclease 9,CRISPR/Cas9)技术建立了uPAR缺陷模型,发现uPAR在具有KRAS突变的肿瘤细胞中可增强MEK/ERK信号转导通路,并通过降解细胞外基质中的整联蛋白和调节EMT影响细胞可塑性[19]。本研究旨在通过建立人胰腺癌类器官和uPAR缺陷或过表达细胞模型,进一步探讨uPAR在自噬(autophagy)过程中对药物响应的调控作用,揭示了uPAR通过调节自噬状态影响细胞对化疗药物敏感性的机制。
1 材料和方法
1.1 患者组织样本
本研究收集了2021年12月—2022年6月在长沙市中心医院(南华大学附属长沙中心医院)行手术切除或活检的8例胰腺肿瘤患者的肿瘤组织样本(由长沙市中心医院中心实验室组织库保存),所有样本经病理学检查确诊为胰腺癌,及时取新鲜肿瘤组织进行了类器官培养,并运用NGS对样本进行了TP53和KRAS突变状态的检测。所有生物学样本的采集和使用均获得长沙市中心医院医学伦理委员会的批准(批准号:2021-S0182,2022-S0084)。
1.2 胰腺癌细胞株
人类胰腺癌细胞株AsPC-1、PANC-1、CAPAN-1、CAPAN-2、MIA PaCa-2和PaTu8988T购自ATCC,置于RPMI-1640培养基含10%胎牛血清、1%L-Glu和1%P/S、37 ℃、CO2体积分数为5%的加湿培养箱中培养。
1.3 基于CRISPR/Cas9系统构建uPAR缺陷细胞模型
采用CRISPR/Cas9技术对uPAR基因进行敲除。根据X-tremeGENE HP DNA转染试剂说明书(德国Merck公司),我们采用两个CRISPR-Cas9载体(美国Sigma-Aldrich公司):pCMV-Cas9-RFP和pCMV-Cas9-GFP共转染细胞。在CRISPR/Cas9瞬时激活后,我们利用荧光活体细胞分类技术筛选出了绿色和红色荧光(green/red fluorescent protein)GFP/RFP双阳性的单个克隆,并进行了培养。随后,采用PCR技术对gRNA的目的位点和可能的缺失位点进行了筛选。
1.4 细胞活力
我们按照说明书使用CellTiter 96® 试剂(美国Promega公司)进行了细胞增殖能力测定。在细胞用0.1 μmol/L吉西他滨处理72 h前,我们首先使用siRNA(80 nmol/L)或抑制剂(JX401:3 µmol/L,3MA:5 µmol/L,CQ:5 µmol/L)预处理细胞24 h。对于类器官,我们将2 000个/孔细胞包埋在96孔板OFM基质胶中,直到形成球形类器官,然后用梯度浓度的吉西他滨(3.9、7.8、15.6、31.5、62.5、125.0和250.0 nmol/L)处理类器官。测量前加入20 μL MTS试剂,37 ℃温育1~3 h,在490 nm和655 nm处测量吸光度(D)值,并计算相对细胞活力或药物半数抑制浓度(half maximal inhibitory concentration,IC50)。
1.5 迁移实验
在细胞生长近100%时,用1%的FCS培养基维持24 h来同步细胞周期,用100 μL无菌移液器吸头在单层细胞上手动划痕,按0、24和48 h拍照并计算相对迁移距离。
1.6 细胞沉默
我们使用了QIAGEN公司提供的RNA和HiPerFect转染试剂,并严格按照包装试剂盒说明书进行了转染实验。简而言之,将含有12 µL HiPerFect、9.6 µL siRNA/阴性对照(80 nmol/ L)和78.4 µL RPMI-1640的100 µL转染培养基在室温下温育20 min,并在接种后立即加入2 mL培养基中的3×105细胞。在进一步处理之前将细胞温育24或48 h。实验所用的siRNA试剂清单见图1F。
1.7 PLAUR基因回补和uPAR再表达
我们按照X-tremeGENE HP DNA转染试剂说明书进行了克隆细胞与人PLAUR ORF克隆载体(pCMV-AC-GFP, NM_002659, Origene)的共转染。通过荧光激活细胞分选术(fluorescence activated cell sorting,FACS)挑选GFP阳性细胞,并使用新霉素G418(400 ng/µL)筛选重新表达uPAR的细胞。uPAR蛋白水平如前所述由酶联免疫吸附实验(enzyme-linked immunosorbent assay,ELISA)或蛋白质印迹法(Western blot)检测确认(图1G、H)。
1.8 抗体和试剂
本实验使用了如下抗体:phospho-p38 MAPK(Thr180/Tyr182;#4511)、phospho-FAK(Tyr397;#8556)、p62(#8025)、LC3B(#3868)、cyclin B1(#12231)、MTHFD2 (#98116)、FoxM1(#5436)、GAPDH(#5174),所有二抗和抗体均购自美国Cell Signal公司;GLUL(#ab176562)和PARK7 (#ab18257)购自英国Abcam公司。吉西他滨(#G5423)和氯喹(#C6628)购自美国Sigma-Aldrich公司,3-MA(#CAY13242)购自德国Biomol公司,p38MAPK抑制物JX401(#2657)购自美国Bio-techne公司。
1.9 ELISA检测
细胞外uPAR蛋白水平检测由ELISA完成(美国R&D公司)。实验前按说明书备好标准品、试剂及待测样本;样本和标准本加入微孔板(至少3个复孔)各50 µL,随后加入50 µL检测液A,37 °C温育1 h,洗板3次。拍板后加检测溶液B 100 µL,37 ℃温育30 min,洗板5次;拍板后加TMB底物90 µL,37 ℃温育10~20 min;最后加终止液50 µL,立即使用酶联免疫检测仪于450 nm处检测D值,绘制标准曲线,并计算标化106个细胞下uPAR的浓度。
1.10 Western blot检测
样品加入放射免疫沉淀法(radio immunoprecipitation assay,RIPA)裂解液,经匀浆、裂解、离心后,获得总蛋白提取液,通过Bradford方法检测提取液中蛋白浓度。采用十二烷基硫酸钠聚丙烯酰胺凝胶电泳(sodium dodecylsulphate polyacrylamide gel electrophoresis,SDS-PAGE)进行蛋白电泳,之后200 mA恒流电1 h将蛋白转印到聚偏二氟乙烯(polyvinylidene fluoride,PVDF)膜上。将PVDF膜放入含5%脱脂奶粉的TBS-T封闭液中封闭1 h,加入一抗,4 ℃摇床过夜。洗膜后加入二抗,37 ℃温育1 h。洗膜后加入发光液,压片,曝光,定影。采用Fusion图像获取和分析系统软件测定蛋白条带的积分光密度值。内部参照按课题组已发表文章中的方法以GAPDH或PARK7蛋白条带作为内参照[20-24]。
1.11 胰腺癌类器官培养
本研究收集的经手术切除的新鲜胰腺肿瘤组织用外科手术刀切碎并在缓冲液 [DMEM、 1%胎牛血清(fetal bovine serum,FBS)、青霉素、链霉素、0.125 mg/mL胶原酶Ⅺ型]中消化并温育2 h。4℃ 条件下 RCF 300×g离心5 min收集后,用Matrigel基质胶重新悬浮细胞并接种在圆形24孔板中。Matrigel基质胶凝固后,加入胰腺类器官培养基置于37 ℃培养箱中培养。胰腺类器官培养基包含DMEM/F12、HEPES 10 mmol/ L、Glutamax 1X、A83-01 500 nmol/ L、mEGF 50 ng/ mL、mNoggin 100 ng/mL、hFGF10 100 ng/ mL、胃泌素Ⅰ 0.01 µmol/L、N-乙酰半胱氨酸1.25 mmol/L、烟酰胺10 mmol/L、B27补充剂、R-spondin条件培养基。通过用含有10.5 µmol/L Y-27632和10 µg/mL DNAseI的胰蛋白酶的消化来传代或冻存类器官。
1.12 统计学处理
计量资料用x±s表示,服从正态分布的计量资料的两组和组间比较采用t检验或方差分析(ANOVA),不服从正态分布时采用非参数检验。细胞生长和耐药性分析采用双因素方差分析(Two-way ANOVA)。相关性分析使用皮尔逊相关系数检验(资料符合正态分布且方差齐性,否则使用Spearman检验)。P<0.05为差异有统计学意义。采用SPSS 26.0和GraphPad Prism 9.0软件进行统计作图。
2 结 果
2.1 uPAR缺陷细胞模型构建结果
本研究以uPAR GFP正引物(外显子4)和uPAR RFP反引物(外显子3)为模板,成功扩增出新的序列片段,表明uPAR外显子3和4之间发生了非同源末端链接(non-homologous end joining,NHEJ)。随后,我们通过聚合酶链式反应(polymerase chain reaction,PCR)和二代测序(next generation sequencing,NGS)技术验证了目标基因的突变情况。uPAR的敲除策略、相关序列引物和基因测序以及uPAR的再表达见图1。
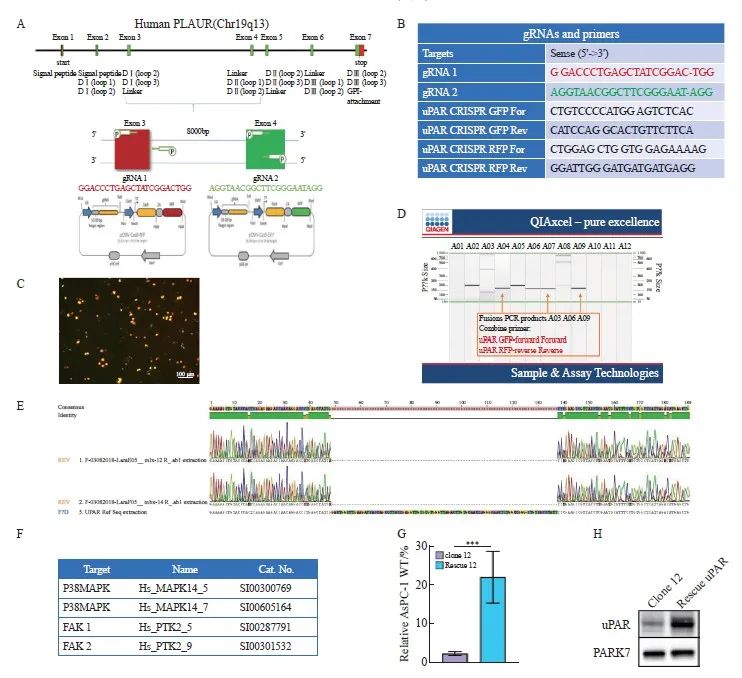
图1 uPAR的敲除策略、相关序列引物和基因测序以及uPAR的再表达
Fig. 1 uPAR knockout strategy and sequencing gene and uPAR re-expression
A: Schematic representation of the uPAR knockout strategy using CRISPR/Cas9. B: List of gRNAs for uPAR and PCR primers used to detect gene knockout. C: Fluorescence-activated cell sorting (FACS) analysis of GFP/RFP double-positive cells. D: PCR screening for gRNA target sites and potential deletions. E: Sanger sequencing of mutations at the gRNA target sites. F: Sequences of siRNA oligonucleotide templates used in this study. G: Re-expression of uPAR protein levels measured by ELISA (n=3, ***: P<0.01, Student’s t-test, two-sided). H: Immunoblot analysis showing de novo expression of uPAR in clone 12 and rescued uPAR cells.
2.2 构建uPAR敲除AsPC-1细胞模型
为了深入探讨uPAR在胰腺癌中的特异性功能,我们构建了uPAR敲除的胰腺癌细胞模型。首先,采用Western blot检测了6种胰腺癌细胞系(AsPC-1、CAPAN-1、CAPAN-2、MIA PaCa-2、PANC-1、PaTu8988T)中uPAR的蛋白水平,结果显示,uPAR在AsPC-1细胞中表达最高(图2A)。进一步通过ELISA检测细胞培养基中的可溶性uPAR(soluble uPAR,suPAR),结果同样显示suPAR在AsPC-1培养基中含量最高 (图2B)。
为构建功能性的uPAR敲除AsPC-1克隆,我们设计了针对uPAR基因外显子3和外显子4的特异性gRNA,并分别标记有绿色和红色荧光蛋白(图1C)。转染细胞72 h后,通过流式细胞术分选获得特定敲除的单克隆细胞。结果如图1D所示,我们以uPAR GFP正引物(外显子4)和uPAR RFP反引物(外显子3)为模板,成功扩增出新的序列片段,表明uPAR外显子3和4之间发生了非同源末端链接(Non-homologous end joining,NHEJ)。最终,我们获得两个纯合子克隆(clone 12和clone 14),其uPAR功能完全缺失。Western blot和ELISA结果确认了克隆细胞中uPAR和suPAR蛋白的缺失(图2C、D)。随后,我们通过PCR和NGS验证了目标基因的突变情况(图1E)。因此,通过成功构建uPAR敲除细胞模型,我们可以进一步探索其在肿瘤生物学中的具体作用。
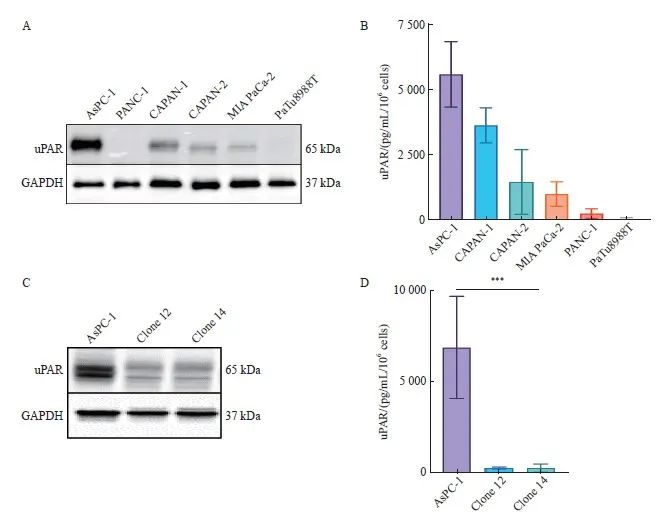
图2 CRISPR-Cas9介导编码uPAR基因敲除的AsPC-1细胞
Fig. 2 CRISPR/Cas9-mediated uPAR knockout in AsPC-1 cells
A: Immunoblot analysis showing uPAR protein levels in the pancreatic cell lines AsPC-1, CAPAN-1, CAPAN-2, MIA PaCa-2, PANC-1, and PaTu8988T. B: ELISA quantification of uPAR protein levels in 6 human pancreatic cell lines. C: Immunoblot analysis comparing uPAR levels between AsPC-1 uPAR knockout (KO) and wild-type (WT) cells. D: ELISA results showing uPAR levels in AsPC-1 uPAR KO cells compared to WT cells. At least three biological replicates were performed. GAPDH was used as a loading control. ***: P<0.001, multiple comparison tests were used to assess differences between groups.
2.3 uPAR的敲除抑制胰腺癌细胞的生长和迁移能力和逆转EMT
已有研究[25]表明uPAR在细胞增殖和运动中起着关键作用。通过MTS实验,我们发现敲除uPAR可显著抑制胰腺癌细胞的生长(P<0.001,图3A)。克隆形成实验进一步证实,敲除uPAR可显著减弱胰腺癌细胞的克隆形成能力(P<0.001,图3B、C)。损伤修复实验结果显示,敲除uPAR可显著延缓胰腺癌细胞的迁移能力(P<0.001,图3D、E)。研究[26]表明,uPAR可以通过与细胞骨架结合并锚定到细胞膜上直接影响细胞迁移。此外,uPAR信号转导功能还可调节细胞可塑性[27]。通过共聚焦荧光显微镜观察,我们发现敲除uPAR后,胰腺癌细胞内F-actin形成显著减少,细胞形态变得更加圆润,细胞膜突起减少(图3F)。这提示uPAR可能通过促进细胞内F-actin的形成增强细胞迁移能力。综上所述,我们的研究结果表明,uPAR在促进胰腺癌细胞的生长、克隆形成和迁移方面起重要作用。
为了进一步探讨uPAR对肿瘤细胞可塑性的影响,本研究采用Western blot进行分析,检测了EMT标志物的表达情况。结果显示,与野生型细胞相比,uPAR敲除的克隆细胞中上皮标志物E-钙黏蛋白(E-cadherin)的表达显著上调,而间质标志物波形蛋白(vimentin)的表达被显著抑制至背景水平(图3G)。此外,转录因子Slug在克隆细胞中的显著表达上调进一步支持uPAR敲除能够逆转EMT表型(图3G)。吉西他滨敏感性实验结果显示,uPAR敲除的克隆细胞对吉西他滨的敏感性显著降低(P<0.001,图3H),提示uPAR可能是影响化疗敏感性的关键因素。综上所述,uPAR在胰腺癌细胞EMT中起到关键作用,其敲除能够逆转EMT表型,且uPAR敲除促使细胞对吉西他滨的耐药性,提示uPAR可能是影响化疗敏感性的标志物。
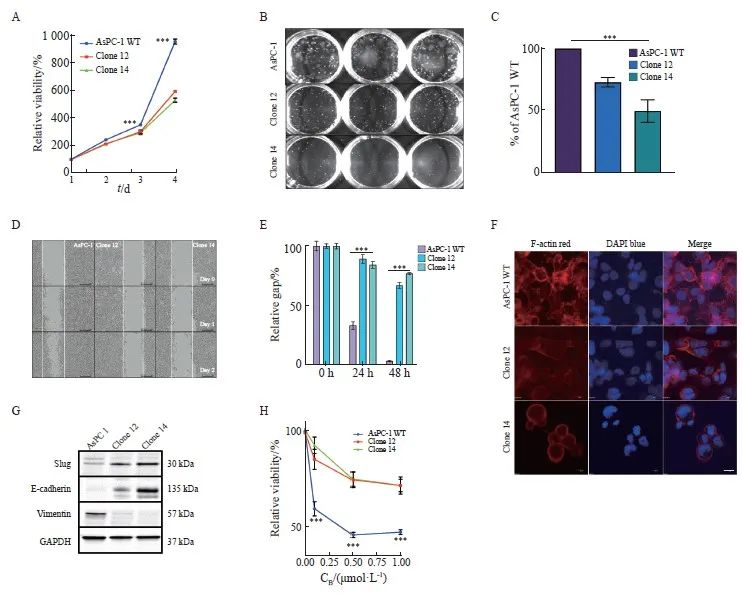
图3 敲除uPAR抑制胰腺癌细胞的生长和迁移能力,并促进吉西他滨化疗耐药性
Fig. 3 uPAR knockout inhibits pancreatic cancer cell growth and migration, and enhances resistance to gemcitabine
A: Growth curves over 4 days showing significantly reduced proliferation rates in AsPC-1 uPAR KO clones compared to WT controls. B: Representative colony formation assay images in methylcellulose comparing uPAR KO and WT cells. C: Quantitative analysis showing a significant reduction in clonogenicity in uPAR KO cells compared to WT cells. D-E: Reduced migratory capacity of uPAR KO cells compared to WT cells. F: Immunofluorescence staining showing fewer stress fibers in uPAR KO cells compared to WT cells (×400, red: phalloidin, blue: DAPI). G: Immunoblot analysis of epithelial and mesenchymal markers indicating Mesenchymal-to-epithelial transition (MET) in uPAR KO cells. H: Increased resistance to gemcitabine in uPAR KO cells (0.1, 0.5, 1 µmol/L, 72 h). At least three biological replicates were performed. GAPDH or PARK7 was used as a loading control. ***: P<0.001, multiple comparison tests were used to assess differences between groups.
2.4 uPAR的敲除激活FAK和p38MAPK的表达和吉西他滨治疗抵抗
研究[11,18]表明,uPAR信号转导涉及FAK和p38MAPK。通过Western blot检测,我们发现uPAR敲除克隆细胞中FAK和p38MAPK的激活水平显著高于对照组(图4A),表明uPAR敲除导致这些信号分子的代偿性激活。肿瘤细胞的化学耐药性与p38MAPK介导的自噬有关[28]。我们通过分析自噬相关蛋白p62和LC3B在克隆细胞中的表达情况,结果发现uPAR敲除克隆细胞中LC3B表达显著上调,而p62蛋白未见显著改变(图4A),提示uPAR可能参与胰腺癌细胞的自噬。已有研究[16]表明,磷酸化FAK促使胰腺癌细胞对吉西他滨产生内在抵抗。为此,我们使用特异性siRNA沉默FAK,结果显示,在uPAR敲除的克隆细胞中,FAK沉默可显著增加吉西他滨敏感性,而在野生型细胞中无此效应(图4B、C)。
2.5 uPAR的敲除激活p38MAPK信号转导并诱导自噬
本研究结果发现,FAK沉默可显著抑制克隆细胞中p38MAPK的激活和LC3B蛋白的表达,但对p62蛋白无明显影响(图4D)。为了探讨p38MAPK介导的自噬是否促进化疗抵抗,我们使用siRNA#1或siRNA#2沉默p38MAPK基因,结果发现p38MAPK的抑制可显著增加克隆细胞对吉西他滨的敏感性(图4E、F)。此外, p38MAPK沉默可显著下调p62和LC3B的表达(图4G),提示p38MAPK的抑制可以显著减少胰腺癌细胞的自噬水平。进一步使用p38的抑制剂JX401取得了类似效果,显著增强了细胞对吉西他滨的敏感性(图4H),提示p38抑制剂JX401联合吉西他滨的治疗策略具有潜力。综上所述,uPAR敲除导致的p38MAPK信号转导通路激活及其引起的自噬增强,揭示了胰腺癌细胞在应对药物压力时的自我保护机制。通过靶向p38MAPK,可以有效地降低自噬水平,从而提高胰腺癌细胞对化疗药物的敏感性。
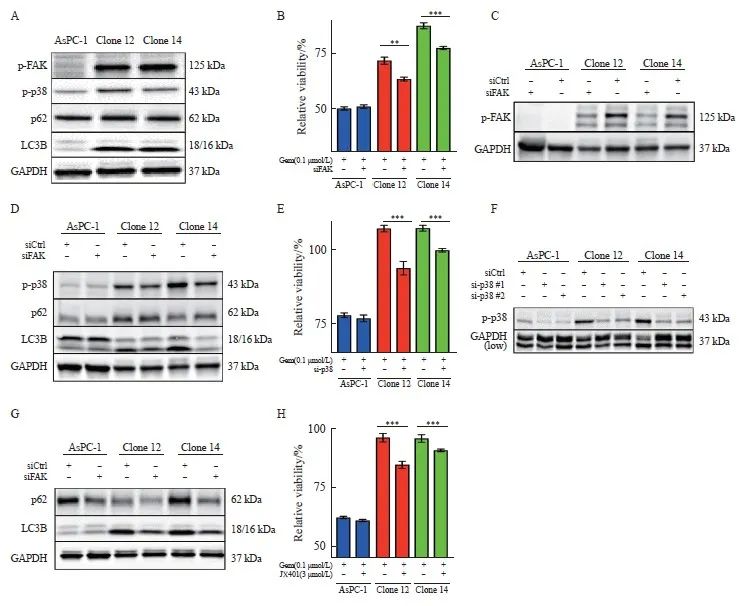
图4 敲除uPAR激活FAK和p38MAPK的表达和促进AsPC-1对吉西他滨化疗抵抗
Fig. 4 uPAR regulates FAK and p38MAPK activity and enhaces gemCitabine resensitivity of AsPC-1 uPAR knockout cells
A: Immunoblot analysis showing increased phosphorylation of FAK and p38, along with elevated levels of LC3B, while p62 levels remain unchanged in uPAR knockout (KO) cells. B-D: Restoration of gemCitabine sensitivity (B) and wild-type (WT) signaling phenotype (D) in AsPC-1 uPAR KO cells after (C) FAK siRNA knockdown. E-F: Gemcitabine response following p38MAPK knockdown (F) using two siRNAs in AsPC-1 uPAR KO cells. G: Re-establishment of the WT signaling phenotype in uPAR KO cells following p38MAPK siRNA knockdown. H: Combined treatment with gemcitabine (0.1 µmol/L) and p38 inhibitor JX401 (3 µmol/L) for 72 h in uPAR KO cells. GAPDH was used as a loading control. At least three biological replicates. **: P <0.01, ***: P<0.001, Student’s t test, two-sided.
2.6 自噬增强AsPC-1细胞对吉西他滨的抗性
自噬在肿瘤细胞存活及治疗抵抗中起关键作用[29]。在我们的实验中,自噬抑制剂3-甲基腺嘌呤(3-methyladenine,3-MA)和氯喹(chloroquine,CQ)的使用降低了自噬标志物LC3B的活性,并部分恢复了克隆细胞对吉西他滨的敏感性,这表明自噬在化疗抵抗中发挥重要作用[30]。为了探讨自噬增强是否导致吉西他滨抗性,我们在AsPC-1克隆细胞中使用3-MA和CQ进行自噬抑制。当两种抑制剂剂量为5 μmol/L时,尽管p62的改变不显著,然而LC3B失活和吉西他滨敏感性恢复在AsPC-1克隆细胞中被显著观察到,而在uPAR野生型细胞中未见到类似效果(图5A、B)。这些结果表明,自噬在AsPC-1细胞中与吉西他滨抗性密切相关,提示自噬抑制剂可能成为克服化疗抵抗的一种潜在治疗策略。
2.7 p38MAPK信号转导和自噬诱导细胞休眠
自噬已被证明在促进休眠肿瘤细胞存活中发挥关键作用[31]。我们的数据显示,uPAR敲除克隆细胞中p38MAPK信号通路激活,以及自噬标志物表达增加,同时伴随着糖酵解标志物GLUL和MTHFD2的表达降低,提示uPAR可能通过这些途径影响癌细胞的休眠状态。因此,我们检测了作为糖酵解标志物的谷氨酸合成酶(glutamate ammonia ligase,GLUL)和亚甲基四氢叶酸脱氢酶2(mitochondrial methylenetetrahydrofolate dehydrogenase 2,MTHFD2),作为p38MAPK诱导休眠和静止的标志物[32-35]细胞周期蛋白B1(cyclin B1,CCNB1)和叉头盒蛋白M1(FoxM1)。Western blot结果显示,与uPAR野生型相比,uPAR敲除克隆中的MTHFD2、FoxM1和CCNB1的表达显著降低(图5C),提示uPAR通过调节自噬和p38MAPK信号通路,抑制胰腺癌细胞进入休眠状态。这一发现揭示了uPAR在肿瘤细胞代谢和休眠状态中的双重调控作用,为胰腺癌的治疗提供了新的靶点。
2.8 uPAR基因回补能部分地恢复吉西他滨敏感性和迁移能力
为了评估uPAR再表达是否可以恢复野生型表型,我们通过PLAUR基因表达载体转染克隆细胞以回补uPAR的再表达(图1G、H)。结果显示,重新表达uPAR可显著抑制p38MAPK和p62蛋白水平(图5D),并恢复对吉西他滨的敏感性(图5E)。此外,uPAR的再表达可显著增强细胞的迁移能力(图5F)。这些结果表明,uPAR在调节胰腺癌细胞对吉西他滨的敏感性和迁移能力中起重要作用,其再表达能够部分地逆转uPAR缺失带来的药物抗性和迁移缺陷。
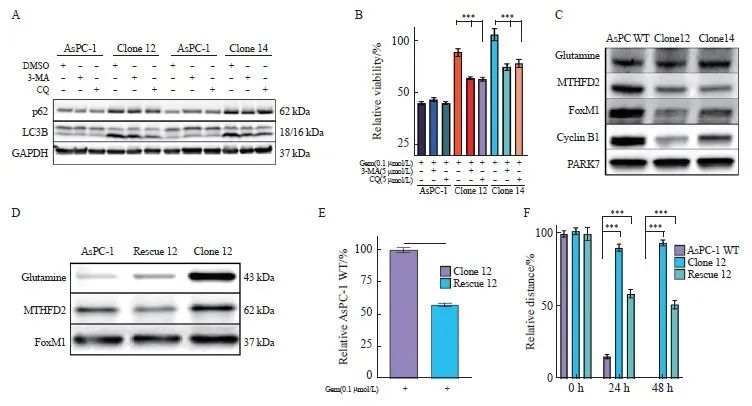
图5 抑制自噬或uPAR再表达使细胞对吉西他滨重新敏感
Fig.5 Inhibition of autophagy or uPAR re-expression restores gemcitabine sensitivity in uPAR knockout cells
A: Immunoblot analysis of autophagy markers p62 and LC3B following treatment with 3-MA (5 µmol/L) or chloroquine (CQ, 5 µmol/L). B: GemCitabine sensitivity after 72 hours of treatment with 3-MA (5 µmol/L) or CQ (5 µmol/L). C: Immunoblot analysis showing altered expression of glycolytic enzymes (GLUL, MTHFD2) and cell cycle regulators (FOXM1, CCNB1), suggesting dormancy in uPAR knockout (KO) cells. D: Re-expression of uPAR in clone 12 cells restored p-p38MAPK and p62 levels to wild-type (WT) levels. E: Re-expression of uPAR restored gemcitabine sensitivity in clone 12 cells (0.1 µmol/L). F: Re-expression of uPAR significantly enhanced migratory capacity in clone 12 cells. GAPDH or PARK7 was used as a loading control. At least three biological replicates were performed. ***: P<0.001, Student’s t-test, two-sided.
2.9 uPAR作为胰腺癌原代类器官对吉西他滨治疗反应的预测生物标志物
胰腺癌患者来源类器官模型(patient-derived organoids,PDO)为临床前治疗评估提供了一种可靠的工具[32]。我们直接从胰腺癌患者样本中培养PDAC类器官(图6A,传代后的类器官培养物形态),并评估uPAR作为吉西他滨疗效预测标志物的价值。通过检测8例胰腺癌类器官培养物对不同浓度吉西他滨的活力情况(图6B),我们计算了吉西他滨的IC50(图6C),并分析了uPAR和自噬标志物LC3B的表达情况(图6D)。结果显示,类器官对吉西他滨的响应与uPAR蛋白水平呈显著负相关(r2=0.66,P <0.05,图6E)。高uPAR水平的PDO对吉西他滨更加敏感,IC50值更小。此外,在低uPAR水平的PDO(n=3)中,自噬抑制剂CQ与吉西他滨联合使用显示出协同抗癌活性,而在高uPAR水平的PDO(n=4)中则未观察到这种协同作用(图 6E、F)。这些结果表明,uPAR可作为胰腺癌吉西他滨治疗反应的预测生物标志物,其表达水平可帮助预测患者对吉西他滨的敏感性,并指导个体化治疗方案的制订,进一步提高胰腺癌的治疗效果。
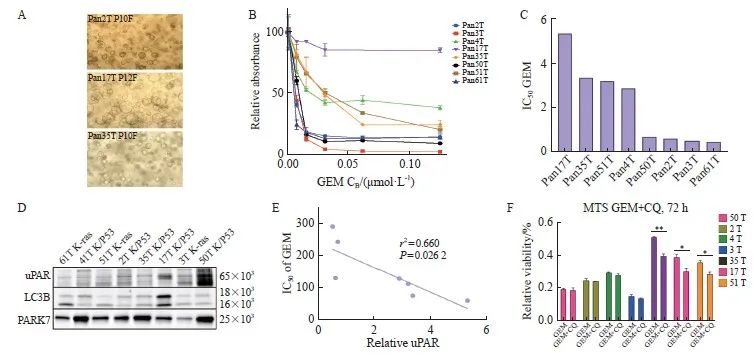
图6 uPAR作为胰腺癌来源的类器官对吉西他滨治疗反应的预测生物标志物
Fig. 6 uPAR as a predictive biomarker for gemcitabine sensitivity in PDAC-derived organoids
A: Representative bright-field images of patient-derived PDAC organoids (PDOs) showing three distinct morphologies: organoids with varying degrees of budding (top), solid organoids (center), and hollow organoids (bottom). Scale bar: 200 µm. B: Dose-response curves for gemcitabine in PDOs cultures (n=8) treated with gemcitabine (0.01-10.00 µmol/L). C: IC50 values for PDOs cultures treated with gemcitabine. D: Immunoblot analysis of uPAR and LC3B in protein extracts from eight PDOs, with PARK7 as a loading control. E: Pearson correlation analysis showing a significant inverse correlation between uPAR levels and gemcitabine sensitivity (IC50) in PDOs (r2=0.66, P=0.026 2). At least three biological replicates were performed. *: P<0.05, compared with each other; **: P<0.01, compared with each other; Student’s t test, two-sided. GEM: Gemcitabine.
3 讨 论
uPAR/PLAUR基因扩增和蛋白水平增加与多种癌症的生物学特征相关,包括非小细胞肺癌和胃癌,通常也是不良预后的标志[33-34]。我们的前期研究结果表明,在胰腺癌患者中,uPAR的扩增与显著缩短的生存期相关[35],特别是在HNF1A阳性的外分泌样肿瘤亚组中[23]。本研究进一步揭示了uPAR在胰腺癌中的关键作用,它能够增强胰腺癌细胞的运动、增殖和肿瘤形成,并通过抑制FAK/p38MAPK信号转导增强胰腺癌细胞对吉西他滨的治疗敏感性。
尽管uPAR的主要功能是通过促进uPA酶反应和ECM的降解来调节细胞行为,但它也具有与细胞外力传递相关的功能,这与uPA酶活性无关[20]。细胞外基质(extracellular matrix,ECM)与整联蛋白的相互作用介导了癌细胞的内在化学耐药性,这种现象也被称为细胞黏附介导的耐药性[36]。细胞黏附介导的耐药性的机制主要包括整联蛋白与ECM的强烈结合,进而激活FAK和p38MAPK信号转导,并诱导细胞进入休眠状态以增加对化学药物的抵抗性。先前的研究[16]表明,FAK的磷酸化介导了AsPC-1细胞对药物的耐药性,而p38MAPK的活化状态对于TP53突变肿瘤对铂类化合物的反应至关重要[37]。此外,自噬对PDAC的生长和发展也起着重要作用[38],该研究[38]还证实自噬的抑制能够阻碍胰腺癌移植瘤的生长和进展。在其他癌症中的研究[28,39]表明,uPAR和纤连蛋白可促进p38MAPK信号转导向RAS-ERK转换,从而促使细胞从休眠状态转向增殖状态[40]。我们的研究显示了,uPAR通过抑制FAK/p38MAPK信号转导诱导胰腺癌细胞的EMT,并增加EMT相关的增殖和迁移。值得注意的是,尽管许多癌细胞在经历EMT后会产生化学耐药[41],但我们观察到表达uPAR的细胞对吉西他滨的敏感性高于uPAR缺陷的细胞。
本研究结果具有潜在的临床意义。我们之前的研究已经表明,uPAR基因扩增水平与胰腺癌患者的不良预后密切相关[35]。我们现在可以解释为什么那些uPAR低表达且p38MAPK活性高的PDAC患者相比于表达uPAR高或uPAR低和p38MAPK低的患者表现更好。此外,我们的发现为制订更有针对性的化疗方案提供了依据:考虑到uPAR高表达患者的预后通常较差,抑制uPAR可能有助于减缓肿瘤生长和转移,并提高肿瘤对化疗的敏感性。另一方面,在uPAR低表达但p38MAPK活性高的肿瘤患者中,联合使用自噬抑制剂(如氯喹)和化疗药物可能会增强治疗效果。早期的临床试验[42-43]已经证明,氯喹与其他细胞毒性药物联合应用可以提高多种实体瘤的治疗效果。
综上所述,本研究揭示了胰腺癌中uPAR扩增的作用机制,即通过下调MAPK/ERK信号转导途径,抑制FAK-p38MAPK介导的EMT、细胞休眠、自噬和化学耐药性。我们的实验还发现,类器官对吉西他滨的响应与uPAR蛋白水平呈负相关。高表达uPAR的胰腺癌类器官对吉西他滨的IC50更低,提示其对该药物有更好的响应。此外,我们在原代胰腺癌来源的7个类器官中观察到3例呈现出协同抗肿瘤活性:自噬抑制剂氯喹联合吉西他滨在低uPAR水平的胰腺癌类器官中显示出协同的抗癌活性,而在高uPAR水平的胰腺癌类器官中未观察到这种协同作用。因此,胰腺癌患者的治疗策略应基于其uPAR和p38MAPK的表达水平进行个体化选择。对于uPAR高表达的患者,uPAR的抑制可能是一种有效的治疗策略;而对于uPAR低表达但p38MAPK活性高的患者,则应考虑联合使用自噬抑制剂和化疗药物。
致谢:本研究得到了德国哥廷根大学附属医院病理科主任Philipp Ströbel教授和Stefan Küffer博士提供的细胞模型以及对实验操作的指导和帮助。
利益冲突声明:所有作者均声明不存在利益冲突。
作者贡献声明:
谭小浪:文章撰写,实施研究过程,数据分析,图片整理;姚莎:实施研究过程,数据分析,参与撰写论文;王桂华:实验分析,文章修改,文献调研与整理;彭罗根:设计研究思路及方案,统计分析,审核文章。
[参考文献]
[1] CONROY T, PFEIFFER P, VILGRAIN V, et al. Pancreatic cancer: ESMO clinical practice guideline for diagnosis, treatment and follow-up[J]. Ann Oncol, 2023, 34(11): 987-1002.
[2] LIN W, NOEL P, BORAZANCI E H, et al. Single-cell transcriptome analysis of tumor and stromal compartments of pancreatic ductal adenocarcinoma primary tumors and metastatic lesions[J]. Genome Med, 2020, 12(1): 80.
[3] PATIL S, STEUBER B, KOPP W, et al. EZH2 regulates pancreatic cancer subtype identity and tumor progression via transcriptional repression of GATA6[J]. Cancer Res, 2020, 80(21): 4620-4632.
[4] NOLL E M, EISEN C, STENZINGER A, et al. CYP3A5 mediates basal and acquired therapy resistance in different subtypes of pancreatic ductal adenocarcinoma[J]. Nat Med, 2016, 22(3): 278-287.
[5] NEVALA-PLAGEMANN C, HIDALGO M, GARRIDOLAGUNA I. From state-of-the-art treatments to novel therapies for advanced-stage pancreatic cancer[J]. Nat Rev Clin Oncol, 2020, 17(2): 108-123.
[6] MIZRAHI J D, SURANA R, VALLE J W, et al. Pancreatic cancer[J]. Lancet, 2020, 395(10242): 2008-2020.
[7] KUMAR A A, BUCKLEY B J, RANSON M. The urokinase plasminogen activation system in pancreatic cancer: prospective diagnostic and therapeutic targets[J]. Biomolecules, 2022, 12(2): 152.
[8] ARONEN A, AITTONIEMI J, HUTTUNEN R, et al. P-suPAR may reflect the inflammatory response after pancreatic surgery[J]. Pancreatology, 2024, 24(1): 146-151.
[9] ZHAI B T, TIAN H, SUN J, et al. Urokinase-type plasminogen activator receptor (uPAR) as a therapeutic target in cancer[J]. J Transl Med, 2022, 20(1): 135.
[10] PESCE N A, PLASTINO F, REYES-GOYA C, et al. Mitigation of human iris angiogenesis through uPAR/LRP-1 interaction antagonism in an organotypic exvivo model[J]. FASEB J, 2024, 38(5): e23533.
[11] SMITH H W, MARSHALL C J. Regulation of cell signalling by uPAR[J]. Nat Rev Mol Cell Biol, 2010, 11(1): 23-36.
[12] FERRARIS G M S, SCHULTE C, BUTTIGLIONE V, et al. The interaction between uPAR and vitronectin triggers ligandindependent adhesion signalling by integrins[J]. EMBO J, 2014, 33(21): 2458-2472.
[13] TAN X M, YAN Y H, SONG B, et al. Focal adhesion kinase: from biological functions to therapeutic strategies[J]. Exp Hematol Oncol, 2023, 12(1): 83.
[14] ZHENG Y H, XIA Y, HAWKE D, et al. FAK phosphorylation by ERK primes ras-induced tyrosine dephosphorylation of FAK mediated by PIN1 and PTP-PEST[J]. Mol Cell, 2009, 35(1): 11-25.
[15] ZHENG Y H, LU Z M. Paradoxical roles of FAK in tumor cell migration and metastasis[J]. Cell Cycle, 2009, 8(21): 3474-3479.
[16] WU H W, LIANG Z Y, SHI X H, et al. Intrinsic chemoresistance to gemcitabine is associated with constitutive and laminininduced phosphorylation of FAK in pancreatic cancer cell lines[J]. Mol Cancer, 2009, 8: 125.
[17] JI C B, ZHANG M L, HU J J, et al. The kinase activity of integrin-linked kinase regulates cellular senescence in gastric cancer[J]. Cell Death Dis, 2022, 13(7): 577.
[18] ZHOU Y H, XIE Y, LUO Y Z, et al. SNAI2 enhances HPVnegative cervical cancer cell dormancy by modulating u-PAR expression and the activity of the ERK/p38 signaling pathway in vitro[J]. Oncol Rep, 2024, 52(2): 104.
[19] HILDENBRAND R, ALLGAYER H, MARX A, et al. Modulators of the urokinase-type plasminogen activation system
for cancer[J]. Expert Opin Investig Drugs, 2010, 19(5): 641-652.
[20] YAO S, PENG L G, ELAKAD O, et al. One carbon metabolism in human lung cancer[J]. Transl Lung Cancer Res, 2021, 10(6): 2523-2538.
[21] BOHNENBERGER H, KADERALI L, STRÖBEL P, et al. Comparative proteomics reveals a diagnostic signature for pulmonary head-and-neck cancermetastasis[J]. EMBO Mol Med, 2018, 10(9): e8428.
[22] ELAKAD O, HÄUPL B, LABITZKY V, et al. Activation of CD44/PAK1/AKT signaling promotes resistance to FGFR1 inhibition in squamous-cell lung cancer[J]. NPJ Precis Oncol, 2022, 6(1): 52.
[23] PENG L G, LI Y C, YAO S, et al. Urokinase-type plasminogen activator receptor (uPAR) cooperates with mutated KRAS in regulating cellular plasticity and gemcitabine response in pancreatic adenocarcinomas[J]. Cancers, 2023, 15(5): 1587.
[24] WIŚNIEWSKI J R, MANN M. A proteomics approach to the protein normalization problem: Selection of unvarying proteins for MS-based proteomics and Western blot[J]. J Proteome Res, 2016, 15(7): 2321-2326.
[25] SHMAKOVA A A, KLIMOVICH P S, RYSENKOVA K D, et al. Urokinase receptor uPAR downregulation in neuroblastoma leads to dormancy, chemoresistance and metastasis[J]. Cancers, 2022, 14(4): 994.
[26] SUGIOKA K, NISHIDA T, KODAMA-TAKAHASHI A, et al. Urokinase-type plasminogen activator negatively regulates α-smooth muscle actin expression via Endo180 and the uPA receptor in corneal fibroblasts[J]. Am J Physiol Cell Physiol, 2022, 323(1): C104-C115.
[27] LOGAN R, JEFFERS A, QIN W Y, et al. TGF-β regulation of the uPA/uPAR axis modulates mesothelial-mesenchymal transition (MesoMT)[J]. Sci Rep, 2021, 11: 21210.
[28] AGUIRRE-GHISO J A, LIU D, MIGNATTI A, et al. Urokinase receptor and fibronectin regulate the ERK (MAPK) to p38 (MAPK) activity ratios that determine carcinoma cell proliferation or dormancy in vivo[J]. Mol Biol Cell, 2001, 12(4): 863-879.
[29] LEVY J M M, TOWERS C G, THORBURN A. Targeting autophagy in cancer[J]. Nat Rev Cancer, 2017, 17(9): 528-542.
[30] BRYANT K L, STALNECKER C A, ZEITOUNI D, et al. Combination of ERK and autophagy inhibition as a treatment approach for pancreatic cancer[J]. Nat Med, 2019, 25(4): 628-640.
[31] LA BELLE FLYNN A, CALHOUN B C, SHARMA A, et al. Autophagy inhibition elicits emergence from metastatic dormancy by inducing and stabilizing Pfkfb3 expression[J]. Nat Commun, 2019, 10(1): 3668.
[32] BOJ S F, HWANG C I, BAKER L A, et al. Organoid models of human and mouse ductal pancreatic cancer[J]. Cell, 2015, 160(1/2): 324-338.
[33] TOKUMO K, MASUDA T, NAKASHIMA T, et al. Association between plasminogen activator inhibitor-1 and osimertinib tolerance in EGFR-mutated lung cancer via epithelialmesenchymal transition[J]. Cancers, 2023, 15(4): 1092.
[34] ZHANG T, WANG B F, SU F, et al. TCF7L2 promotes anoikis resistance and metastasis of gastric cancer by transcriptionally activating PLAUR[J]. Int J Biol Sci, 2022, 18(11): 4560-4577.
[35] HILDENBRAND R, NIEDERGETHMANN M, MARX A, et al. Amplification of the urokinase-type plasminogen activator receptor (uPAR) gene in ductal pancreatic carcinomas identifies a clinically high-risk group[J]. Am J Pathol, 2009, 174(6): 2246-2253.
[36] HENKE E, NANDIGAMA R, ERGÜN S. Extracellular matrix in the tumor microenvironment and its impact on cancer therapy[J]. Front Mol Biosci, 2019, 6: 160.
[37] HUANG J Y, LIN Y C, CHEN H M, et al. Adenine combined with cisplatin promotes anticancer activity against hepatocellular cancer cells through AMPK-mediated p53/p21 and p38 MAPK cascades[J]. Pharmaceuticals, 2022, 15(7): 795.
[38] YAMAMOTO K, VENIDA A, YANO J, et al. Autophagy promotes immune evasion of pancreatic cancer by degrading MHC-I[J]. Nature, 2020, 581(7806): 100-105.
[39] GANDHARI M, ARENS N, MAJETY M, et al. Urokinase-type plasminogen activator induces proliferation in breast cancer cells[J]. Int J Oncol, 2006, 28(6): 1463-1470.
[40] GONIAS S L, HU J J. Urokinase receptor and resistance to targeted anticancer agents[J]. Front Pharmacol, 2015, 6: 154.
[41] SHIBUE T, WEINBERG R A. EMT, CSCs, and drug resistance: the mechanistic link and clinical implications[J]. Nat Rev Clin Oncol, 2017, 14(10): 611-629.
[42] MEHNERT J M, MITCHELL T C, HUANG A C, et al. BAMM (BRAF autophagy and MEK inhibition in melanoma): a phase Ⅰ/Ⅱ trial of dabrafenib, trametinib, and hydroxychloroquine in advanced BRAFV600-mutant melanoma[J]. Clin Cancer Res, 2022, 28(6): 1098-1106.
[43] JAIN V, HARPER S L, VERSACE A M, et al. Targeting UGCG overcomes resistance to lysosomal autophagy inhibition[J]. Cancer Discov, 2023, 13(2): 454-473.
- 搜索
-
- 1000℃Nutrients:真实世界数据,纤维肌痛患者的饮食与运动自适应规律
- 1000℃D-二聚体升高诊治与管理专家共识(2026)
- 1000℃专家论坛|文良志:门静脉血栓的诊断和治疗
- 1000℃首例儿童NF2驱动型胸膜间皮瘤,多方法学检测锁定NF2双等位基因失活和14/22号染色体缺失,提示与成人胸膜间皮瘤不同
- 1000℃打破误区:干扰素追求CHB功能性治愈,HBsAg为何“不降反增”?
- 1000℃迷惑性极强的肝内病灶!影像表现疑点重重,最终病理竟查出两种不同肝脏恶性肿瘤
- 1000℃指南共识|原发性肝癌分子靶向药物相关蛋白尿中西医结合诊疗专家共识
- 1000℃Diabetologia:意大利北部社区 1~100 岁人群胰岛自身抗体与乳糜泻 TGA-IgA 的年龄分布及检测方法学验证
- 精J Child Psychol Psychiatry:12种罕见神经发育障碍儿童沟通能力谱系
- 精研究发现:爱吃辣的人,心血管病和癌症死亡风险都会显著降低
- 精Nursing in Critical Care:别再指责护士了!ICU 里被遗漏的护理,根源在系统而非个人
- 精Acta Obstet Gynecol Scand:罕见病女性的妊娠并发症与母婴结局,一项单中心434种罕见病的回顾性队列研究
- 精【爱儿小醉】儿科患者术前对流层臭氧暴露与围手术期呼吸系统不良事件之间的关系:一项单中心回顾性队列研究
- 精eBioMedicine:牙龈下微生物组与脑健康存在连续关联梯度,牙周炎或成认知衰退可干预靶点
- 精军事医学研究院《自然·通讯》:自适应IrPtCu纳米酶水凝胶实现耐药菌感染伤口序贯治疗
- 精能够逆转萎缩性胃炎的两个中成药,该怎么选择?
- 荐同时性多发性原发性肺癌,左右病灶分别为EGFR和ALK阳性,考虑淋巴结肿大仅局限左肺门及血浆EGFR阳性,采用奥希替尼联合化疗
- 荐40岁女性同时罹患卵巢支持细胞-间质细胞瘤和透明细胞乳头状肾肿瘤,WES等基因检测竟为阴性
- 荐椎管内麻醉使用止血药突发气道痉挛的抢救流程解析
- 荐女子肝区无任何不适,影像提示复杂囊性病变,层层鉴别后锁定罕见胆管源性囊性肿瘤
- 荐“绘”真报告 | 病理考虑为中枢神经细胞瘤,检出脑室外神经细胞瘤的特征性变异FGFR1-TACC1融合,辅助鉴别诊断
- 荐8岁女童出现男性化症状,竟是形似「性索-间质肿瘤」的卵巢「无性细胞瘤」所致,少见KRAS/CDK4共扩增或与侵袭性有关
- 荐17例病例分析揭示常见于中年人的色素性室管膜瘤临床特征与预后,分子检测可助力临床精准诊疗
- 荐Lancet Oncol:结直肠癌腹膜转移,围手术期化疗并非必选项
- 标签列表
-
- 星座 (702)
- 孩子 (526)
- 恋爱 (505)
- 婴儿车 (390)
- 宝宝 (328)
- 狮子座 (313)
- 金牛座 (313)
- 摩羯座 (302)
- 白羊座 (301)
- 天蝎座 (294)
- 巨蟹座 (289)
- 双子座 (289)
- 处女座 (285)
- 天秤座 (276)
- 双鱼座 (268)
- 婴儿 (265)
- 水瓶座 (260)
- 射手座 (239)
- 不完美妈妈 (173)
- 跳槽那些事儿 (168)
- baby (140)
- 女婴 (132)
- 生肖 (129)
- 女儿 (129)
- 民警 (127)
- 狮子 (105)
- NBA (101)
- 家长 (97)
- 怀孕 (95)
- 儿童 (93)
- 交警 (89)
- 孕妇 (77)
- 儿子 (75)
- Angelababy (74)
- 父母 (74)
- 幼儿园 (73)
- 医院 (69)
- 童车 (66)
- 女子 (60)
- 郑州 (58)