首页 > 医疗资讯/ 正文
引言
在现代生物医学研究中,了解基因调控机制对于揭示生命过程和疾病发生的本质至关重要。尽管编码区(即基因)的研究已取得了显著进展,但非编码区域,占据了人类基因组大部分的区域,其功能和调控机制仍然是一个相对较少被理解的领域。非编码区域包含了大量的顺式调控元件(cis-regulatory elements, CREs),如启动子、增强子和沉默子,它们在基因表达调控中扮演着不可或缺的角色。因此,精确地鉴定和功能性地验证这些元件是理解基因调控网络的关键一步。
随着CRISPR技术的快速发展,CRISPR干扰(CRISPR interference, CRISPRi)已成为研究非编码区域功能的有力工具。CRISPRi利用dCas9蛋白质和引导RNA(sgRNA)的系统,通过靶向特定的DNA序列来抑制附近基因的表达,而不改变DNA序列。这种方法的优势在于其高度的特异性和可逆性,使其成为功能性基因组学研究中不可或缺的工具。
非编码基因组的研究揭示了其在调节基因表达、维持细胞状态和参与疾病过程中的关键作用。通过CRISPRi筛选,研究人员可以系统地研究非编码区域如何影响特定基因的表达,进而揭示基因调控网络中的新机制和潜在的治疗靶点。
ENCODE(Encyclopedia of DNA Elements)计划旨在编目和注释人类基因组中的所有功能性元素,特别是非编码区域。通过整合大规模的基因组学数据,ENCODE计划已经成功地标注了成千上万的顺式调控元件,为非编码区域的功能研究提供了宝贵的资源。ENCODE项目的多中心综合分析揭示了非编码CRISPRi筛选的潜力,为未来CRISPRi筛选设计和分析提供了实验指南,加速了非编码基因组功能特性的研究。(3月19日 Nature Methods “Multicenter integrated analysis of noncoding CRISPRi screens”)

Highlights
该研究通过多中心整合分析非编码CRISPR干扰筛选(noncoding CRISPRi screens),推动了我们对基因调控景观的理解。ENCODE (Encyclopedia of DNA Elements)联盟的努力在注释非编码顺式调控元件(cis-regulatory elements, CREs)方面取得了进展。研究使用超过540,000次的扰动(perturbations),覆盖了近24.85兆碱基对的基因组,揭示了332个功能性确认的CRE-基因联系(CRE–gene links)。通过在K562细胞中进行的分析,研究确立了利用CRISPR干扰(CRISPR interference, CRISPRi)筛选内源性非编码元件的指南,包括准确检测显示变化且通常效应低的CREs的能力。
此外,研究通过比较五种筛选分析工具(benchmarking five screen analysis tools),发现CASA在CRE调用方面最为保守,且能有效抵御低特异性sgRNA引起的假象。
研究还发现CRISPRi在转录区域(transcribed regions)存在微妙的DNA链偏见(strand bias),这对筛选设计和分析具有重要意义。
这项研究提供了一个易于访问的数据资源,预设计的sgRNA用于目标3,275,697个ENCODE SCREEN候选CREs的CRISPRi和筛选指南,加速了非编码基因组的功能特征研究。
Strategies
该研究利用了一种系统性方法,通过混合的非编码CRISPR筛选(pooled noncoding CRISPR screens)来研究顺式调控机制(cis-regulatory mechanisms)。ENCODE4 功能性特征中心(Functional Characterization Centers)执行了108次筛选,在人类细胞系中进行了超过540,000次的基因组扰动(perturbations),涵盖了24.85兆碱基对。这些筛选依靠CRISPR干扰技术(CRISPR interference, CRISPRi),一个利用CRISPR/Cas9系统定向抑制基因表达的方法。研究使用了332个在K562细胞中功能性确认的CRE-基因联系(CRE–gene links),建立了筛选内源性非编码元素的准则。
CRISPRi筛选设计: 利用CRISPRi系统,研究人员设计了针对特定非编码DNA区域的sgRNA,以系统性地探索这些区域对基因表达的调控作用。这些sgRNAs特定靶向被认为是可能的调控元件,如增强子(enhancers)或沉默子(silencers)。
数据分析工具的基准测试: 研究比较了五种不同的筛选分析工具,包括CASA,来评估哪种工具在确定CRE时最为保守且能有效规避低特异性sgRNA带来的问题。
功能性确认的CRE-基因联系: 通过在特定的细胞类型(如K562细胞)中进行大规模的CRISPRi筛选,研究人员能够识别并确认CRE与特定基因之间的直接联系。
设计和分析的技术建议: 根据对不同CRISPR筛选方法的比较和分析,研究提供了针对如何设计和执行CRISPRi筛选的技术建议,以及如何解析结果以加速非编码基因组的功能性特征研究。
提供数据资源和预设计sgRNAs: 研究提供了针对ENCODE SCREEN候选CREs的3,275,697个预设计的sgRNAs,这是一个为相关领域研究人员提供的重要资源,可以直接用于未来的功能性筛选实验。
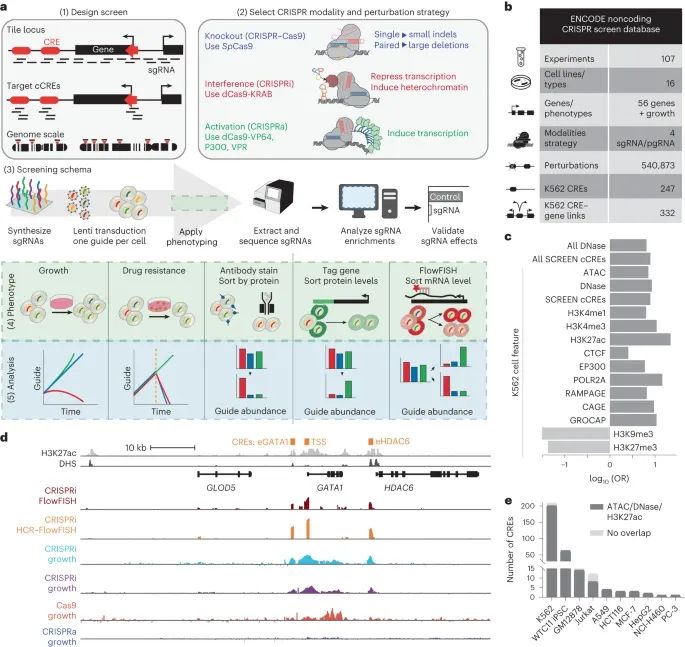
ENCODE 非编码 CRISPR 筛选数据库的概况。(Credit: Nature Methods)
a. 非编码 CRISPR 筛选的五个主要策略:(1) 干预设计策略;(2) CRISPR 操作方式和干预策略;(3) 标准筛选的工作流程;(4) 表型策略;以及 (5) 分析方法。这部分还提到了使用的 CRISPR 系统,如 SpCas9 (来自 Streptococcus pyogenes 的 Cas9) 以及所引发的插入和缺失 (indels)。
b. 概述了 2022 年 4 月 ENCODE 门户发布的人类细胞系/类型中进行的 CRISPR 筛选数据。这里列出了实验、细胞系/类型、操作方式、策略、基因/表型和干预措施等各项数据,并特别提到了 K562 细胞系中的 CREs (顺式调控元素) 和 CRE-基因联系,使用了配对的 sgRNA (pgRNA)。
c. 提供了基因组注释与 CRISPR 筛选识别的调控元素之间重叠的比例 (OR,优势比) 数据,包括 210 个案例。这里区分了适用于所有细胞类型的特征(All)和特定于 K562 细胞类型的注释。所有的 OR 值在 P < 0.01 的水平上均显著,且为了便于查看,这些值被转换为了对数。
d. 展示了 K562 细胞中 GATA1 位点的基因组浏览器快照,包括 H3K27ac (浅灰色) 和 DHS 信号 (深灰色)。同时显示了一次 CRISPR 筛选数据的信号对数变化 (log2(FC)),涵盖了 CRISPRi FlowFISH、CRISPRi HCR-FlowFISH、以及其他几种不同 CRISPR 策略的数据。之前验证的 GATA1 CREs 在图顶部以橙色标出。
e. 显示在 CRISPR 筛选中具有显著性的 CREs 数量,这些 CREs 与通过 ATAC-seq 和 DNase-seq 定义的染色质可及性区域重叠,以及/或 H3K27ac ChIP-seq 峰值(深灰色)或在十个细胞线中不重叠这些特征。数据显示了不同细胞线中的重叠情况,如 A549、GM12878、HCT116 等,突出了 K562 细胞线中 200/210 的显著 CREs 与已知的染色质可及性区域重叠。
Behind the Scenes
非编码原件筛选的设计
非编码原件在基因调控中扮演着至关重要的角色。为了准确揭示它们在基因表达和疾病发展中的作用,设计高效且精确的CRISPRi筛选实验至关重要。
针对转录区域的CRISPRi筛选设计
目标选择:选择合适的非编码区域是实验设计的第一步。建议优先考虑已知或预测与疾病相关性较高的区域,如转录起始位点附近的增强子或沉默子。
sgRNA设计:设计针对目标非编码元素的sgRNA时,需确保其具有高特异性和高编辑效率。利用最新的算法和数据库进行设计,避免潜在的脱靶效应。
实验验证:在正式筛选前,对一小部分sgRNA进行预实验,以验证其在细胞中的编辑效率和特异性。
DNA链偏好性的考虑
CRISPRi技术在特定区域的编辑效率可能会受到DNA链偏好性的影响。研究表明,针对模板链的sgRNA可能会因为RNA聚合酶的存在而导致编辑效率降低。因此,在设计实验时,以下几点值得注意:
双链考虑:设计针对目标非编码元素的sgRNA时,应同时考虑针对正链和负链的设计,以比较不同链上sgRNA的编辑效率。
实验设计:通过设置对照组来明确DNA链偏好性对CRISPRi编辑效率的影响。例如,可以将针对正负链的sgRNA在相同条件下对照实验,以评估其效率差异。
数据分析:在分析筛选结果时,应考虑DNA链偏好性对数据的可能影响,合理解释实验结果,以避免偏差。
数据资源和预设计sgRNA
为目标ENCODE SCREEN候选CREs的sgRNA
ENCODE数据库的应用:ENCODE(Encyclopedia of DNA Elements)项目提供了一个全面的数据集,涵盖了人类基因组中的非编码区域。通过利用这些数据,研究人员可以筛选出与特定生物学过程或疾病状态相关的候选CREs。
预设计sgRNA的选择:基于ENCODE项目数据,已经开发出了多个工具和资源库,提供预设计的针对特定非编码区域的sgRNA。这些资源使研究人员能够直接选择高效、特异性强的sgRNA,省去了设计和验证的时间。
加速非编码基因组功能性特征研究的指南
资源的整合应用:
利用ENCODE项目和其他公开数据库(如UCSC Genome Browser, GTEx)识别功能性猜测较高的非编码区域。
通过CRISPRi设计软件和数据库(如CRISPOR, Benchling)选择或验证sgRNA的特异性和效率。
实验设计的优化:
结合转录组学和表观遗传学数据,优先考虑那些在特定细胞类型或疾病条件下表达差异显著的非编码区域。
应用机器学习方法对大量sgRNA效率数据进行分析,进一步优化sgRNA设计。
数据分析与验证:
利用高通量测序技术(如RNA-seq)分析CRISPRi编辑后的基因表达变化,确定非编码元素的功能。
通过基因表达和表观遗传标记的改变,验证CREs对相应基因调控的影响。
筛选分析工具的测试
在这项多中心整合分析的非编码CRISPRi筛选研究中,研究人员基准测试了五种筛选分析工具,旨在确定哪种工具在鉴定cis-调控元件(CREs)时最为有效和可靠。
经过比较,CASA(CRISPR Analytical Suite Application)被发现产生最保守的CRE调用。这意味着,与其他四种工具相比,CASA在确定一个给定的基因区域作为CRE时更加谨慎,减少了假阳性的产生,从而提高了CRE调用的准确性。
此外,CASA对于低特异性sgRNA的人为误差表现出了较强的鲁棒性。低特异性sgRNA可能导致CRISPRi实验中的非特异性编辑,这些编辑可能会影响实验结果的解释。CASA能够有效识别并减少这类低特异性引导RNA带来的潜在误差,保证筛选结果的可靠性。
CRE调用数量:与其他四种分析工具(aggrDESeq2, CRISPR-SURF, MAGeCK, 和 RELICS)相比,CASA鉴定出的CRE数量最少。这表明CASA在鉴定CRE时更为保守,只有信号最强、数据最为确凿的区域才会被识别为CRE,从而减少假阳性的产生。
与已知注释的重叠:CASA调用的CRE与已知的ENCODE标注的CREs、H3K27ac峰和DHS(DNase I敏感位点)的重叠比例较高。这意味着CASA鉴定的CREs与已经通过其他生物学实验证实具有调控活性的区域有较高的一致性,进一步说明了其CRE调用的准确性和保守性。
对低特异性sgRNA的鲁棒性:在包含低特异性sgRNA的CRISPRi筛选数据分析中,CASA与其它方法相比,其识别出的CRE数量不受低特异性sgRNA的影响,数量保持不变。这表明CASA能有效排除因低特异性sgRNA导致的非特异性编辑,从而避免了假阳性CRE的识别,保证了结果的鲁棒性。
重复性实验的一致性:在使用CASA分析不同生物学重复实验的数据时,得到的CRE调用结果显示高度一致性。这种一致性说明CASA能够可靠地识别真正的CREs,即便在实验条件或操作过程中存在微小变化时也能保持稳定的结果输出。
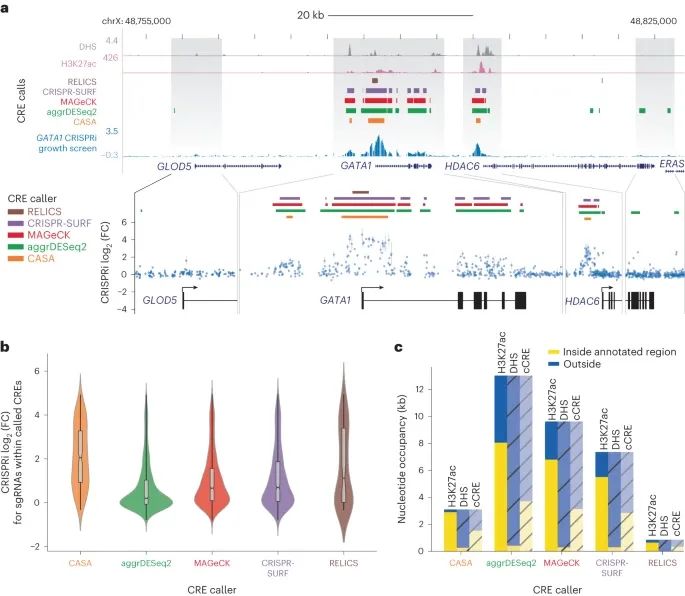
不同的 CRISPR 筛选分析工具在识别 CREs(顺式调控元素)时具有不同的选择性。(Credit: Nature Methods)
a. 部分展示了在 GATA1 位点进行的 CRISPRi 生长筛选中,sgRNA介导的生长影响(蓝色)、H3K27ac ChIP信号(粉红色)和DHS(灰色)。为了减少可能导致混淆的非目标毒性,使用 GuideScan 聚合的 CFD 分数小于 0.2 的低特异性 sgRNAs 被过滤掉。不同的CRISPR筛选分析工具,如 CASA(橙色)、aggrDESeq2(绿色)、MAGeCK(红色)、CRISPR-SURF(紫色)和 RELICS(棕色),的峰值调用显示在密集的轨迹中。放大的区域展示了单个sgRNA效应的对数变化(log2(FC)),点代表两次重复实验的平均值,条形表示观察值的最小-最大范围。
b. 展示了不同 CRISPR 筛选分析工具识别的峰内 sgRNAs 的平均引导效应分布。这里使用了箱形图和小提琴图来展示数据的分布情况,中心线代表中位数,凹口代表中位数的置信区间,盒子的界限代表第一和第三四分位数,而须线展示了所有数据点的范围。sgRNA 的数量从左到右分别是 204、1,218、715、623 和 71。显示了不同工具在鉴定 CREs 时的选择性和敏感性。
c. 部分展示了 CRISPRi 筛选峰区与已注释的染色质特征(H3K27ac、DHS)和 ENCODE SCREEN cCREs 的重叠(黄色)与补充(蓝色)情况。着色和阴影表示用于比较的参考注释,而总条高度反映了各峰值调用工具标记为显著的总基因组区域。这部分强调了不同分析工具在定义 CREs 的显著性和其与已知染色质特征重叠度方面的差异,展现了不同工具在综合数据分析和解释基因调控复杂性方面的独特优势和局限。
CRISPRi在转录区域展现出的微妙DNA链偏好性
CRISPRi效应与DNA链的关系:在对GATA1基因体(gene body)进行CRISPRi筛选时,研究人员观察到,针对编码链(coding strand)的sgRNA对细胞生长的影响显著大于针对模板链(template strand)的sgRNA。这种差异表明,CRISPRi对基因表达的抑制作用在不同DNA链上有着不同的效力,其中针对编码链的sgRNA更能有效地抑制目标基因的表达。
基因内sgRNA效应的分布:进一步的分析显示,在GATA1基因内,针对编码链的sgRNA在整个转录区域内均显示出较强的抑制效应,而这种效应在转录终止位点(TES)之后急剧下降。这种效应分布模式进一步证实了CRISPRi在转录区域的DNA链偏好性。
CRISPRi与CRISPR激活(CRISPRa)效应的比较:研究中还比较了CRISPRi和CRISPRa(一种用于增强目标基因表达的CRISPR技术)在相同基因体区域的效应。结果显示,与CRISPRi显示出的链特异性不同,CRISPRa在编码链和模板链上的激活效果更为一致。这种对比进一步突出了CRISPRi特有的链偏好性。
不同CRISPR技术的效应对比:通过对CRISPRi、CRISPRa和dCas9(一种不具切割活性的Cas9变体)的比较,发现CRISPRi在基因体内显示出的链偏好性是与KRAB(一种转录抑制因子)的耦合作用相关的。相比之下,单独使用dCas9时,这种链偏好性不显著,说明CRISPRi特有的链偏好性与其所用的抑制因子有关。
功能性CRE–基因联系的确认
在对非编码区域的研究中,CRE(顺式调控元件)与特定基因之间的功能性连接的确认,是理解基因表达调控机制的关键一步。通过ENCODE项目的深入研究与CRISPRi技术的应用,研究人员在K562细胞中成功确认了332个功能性的CRE–基因连接,这一成果标志着非编码基因组研究的一个重要进展。
在K562细胞中确认的332个功能性CRE–基因联系
K562细胞作为一种白血病细胞系,因其遗传背景的稳定性和易于操作的特点,成为非编码基因组研究的理想模型。通过对108次CRISPRi筛选实验的数据分析,研究团队不仅揭示了数百个先前未知的CREs,更重要的是,确认了它们与特定基因之间的直接功能性连接。这些功能性连接的确认,不仅加深了我们对基因调控网络的理解,也为后续的疾病机理研究和治疗目标的开发提供了宝贵的线索。
CRISPRi筛选中CRE的准确检测
CRISPRi技术在非编码区域研究中的应用,使得对CREs的功能性分析成为可能。通过设计特异性高的sgRNA靶向特定非编码区域,CRISPRi能够有效地抑制这些区域的活性,进而观察对应基因表达的变化。这一方法的高效性和精确性,为CRE的功能性验证提供了强有力的技术支持。
非编码CRISPRi筛选在基因调控研究中的意义
对基因调控机制理解的贡献
非编码CRISPRi筛选技术使研究人员能够精确地靶向和调节基因组中的非编码区域,揭示了这些区域在基因表达调控中的重要作用。通过这一技术,研究人员已经能够识别出多个与特定生物学过程和疾病状态相关的功能性顺式调控元件(CREs),这对于解码生命复杂性提供了新的视角。
未来方向和挑战
尽管非编码CRISPRi筛选技术已取得显著成就,但在未来的研究中仍面临着若干挑战和发展方向:
技术精确性的提升:随着研究的深入,提高CRISPRi系统的靶向精度和编辑效率成为迫切需要解决的问题。这包括开发新的靶向策略和优化现有系统,以减少非特异性编辑和提高编辑成功率。
数据集的扩充与整合:建立更全面的非编码区域功能数据库,整合跨种属的数据资源,对于深入理解非编码区域在不同生物学背景下的功能至关重要。
疾病模型的开发:利用非编码CRISPRi筛选技术构建更为精确的疾病模型,以模拟人类疾病的复杂性和多样性,为疾病机理研究和新药开发提供强大支持。
伦理和安全性的考量:随着技术的应用越来越广泛,其伦理和安全性问题也越来越受到关注。确保科技发展的同时,保护个人隐私和生物伦理成为不可忽视的重要议题。
Q&A
非编码CRISPRi筛选是如何帮助识别功能性CREs的?
非编码CRISPRi(CRISPR干扰)筛选技术,通过特异性地抑制基因组中的非编码区域表达,帮助研究人员识别那些对基因表达调控至关重要的功能性顺式调控元件(CREs)。这一过程包括选定目标非编码区域、设计特异性的CRISPRi系统,以及评估目标抑制对细胞表型或基因表达的影响。此方法能够揭示非编码区域与特定基因或生物学路径之间的直接关联,从而为理解基因调控网络提供了强有力的工具。
在非编码CRISPRi筛选中,如何选择目标区域?
选择非编码CRISPRi筛选的目标区域,通常依据生物信息学预测、已知的功能性元件数据库,以及与疾病相关性的研究结果。首先,研究人员会利用高通量测序技术,如ChIP-seq(染色质免疫沉淀测序)和ATAC-seq(染色质可及性测序),来识别潜在的调控元件。接着,通过比对相关疾病或生物学过程中的表达谱,筛选出具有潜在重要生物学功能的非编码区域。此外,考虑到非编码区域在不同细胞类型或生物学条件下可能具有不同的功能,选择时还需考虑实验背景和研究目标。
筛选分析工具CASA与其他工具相比有何优势?
CASA(CRISPRi筛选分析软件)是一款专为非编码CRISPRi筛选数据分析而设计的软件工具,它在多个方面显示出相对于其他分析工具的优势。首先,CASA能够处理大规模筛选数据,提供快速准确的分析结果。其次,CASA具有高度的灵活性,可以适应不同实验设计和数据类型,包括针对单个或多个基因的筛选。此外,CASA在识别功能性CREs方面显示出较高的特异性和灵敏度,尤其是在分析低特异性sgRNA数据时。这使得CASA成为在复杂基因组背景下进行精确功能性元件识别的强大工具。
原文链接
https://www.nature.com/articles/s41592-024-02216-7
Yao D, Tycko J, Oh JW, et al. Multicenter integrated analysis of noncoding CRISPRi screens. Nat Methods. Published online March 19, 2024. doi:10.1038/s41592-024-02216-7
- 搜索
-
- 1000℃Nutrients:真实世界数据,纤维肌痛患者的饮食与运动自适应规律
- 1000℃D-二聚体升高诊治与管理专家共识(2026)
- 1000℃专家论坛|文良志:门静脉血栓的诊断和治疗
- 1000℃首例儿童NF2驱动型胸膜间皮瘤,多方法学检测锁定NF2双等位基因失活和14/22号染色体缺失,提示与成人胸膜间皮瘤不同
- 1000℃打破误区:干扰素追求CHB功能性治愈,HBsAg为何“不降反增”?
- 1000℃迷惑性极强的肝内病灶!影像表现疑点重重,最终病理竟查出两种不同肝脏恶性肿瘤
- 1000℃指南共识|原发性肝癌分子靶向药物相关蛋白尿中西医结合诊疗专家共识
- 1000℃Diabetologia:意大利北部社区 1~100 岁人群胰岛自身抗体与乳糜泻 TGA-IgA 的年龄分布及检测方法学验证
- 精J Child Psychol Psychiatry:12种罕见神经发育障碍儿童沟通能力谱系
- 精研究发现:爱吃辣的人,心血管病和癌症死亡风险都会显著降低
- 精Nursing in Critical Care:别再指责护士了!ICU 里被遗漏的护理,根源在系统而非个人
- 精Acta Obstet Gynecol Scand:罕见病女性的妊娠并发症与母婴结局,一项单中心434种罕见病的回顾性队列研究
- 精【爱儿小醉】儿科患者术前对流层臭氧暴露与围手术期呼吸系统不良事件之间的关系:一项单中心回顾性队列研究
- 精eBioMedicine:牙龈下微生物组与脑健康存在连续关联梯度,牙周炎或成认知衰退可干预靶点
- 精军事医学研究院《自然·通讯》:自适应IrPtCu纳米酶水凝胶实现耐药菌感染伤口序贯治疗
- 精能够逆转萎缩性胃炎的两个中成药,该怎么选择?
- 荐同时性多发性原发性肺癌,左右病灶分别为EGFR和ALK阳性,考虑淋巴结肿大仅局限左肺门及血浆EGFR阳性,采用奥希替尼联合化疗
- 荐40岁女性同时罹患卵巢支持细胞-间质细胞瘤和透明细胞乳头状肾肿瘤,WES等基因检测竟为阴性
- 荐椎管内麻醉使用止血药突发气道痉挛的抢救流程解析
- 荐女子肝区无任何不适,影像提示复杂囊性病变,层层鉴别后锁定罕见胆管源性囊性肿瘤
- 荐“绘”真报告 | 病理考虑为中枢神经细胞瘤,检出脑室外神经细胞瘤的特征性变异FGFR1-TACC1融合,辅助鉴别诊断
- 荐8岁女童出现男性化症状,竟是形似「性索-间质肿瘤」的卵巢「无性细胞瘤」所致,少见KRAS/CDK4共扩增或与侵袭性有关
- 荐17例病例分析揭示常见于中年人的色素性室管膜瘤临床特征与预后,分子检测可助力临床精准诊疗
- 荐Lancet Oncol:结直肠癌腹膜转移,围手术期化疗并非必选项
- 标签列表
-
- 星座 (702)
- 孩子 (526)
- 恋爱 (505)
- 婴儿车 (390)
- 宝宝 (328)
- 狮子座 (313)
- 金牛座 (313)
- 摩羯座 (302)
- 白羊座 (301)
- 天蝎座 (294)
- 巨蟹座 (289)
- 双子座 (289)
- 处女座 (285)
- 天秤座 (276)
- 双鱼座 (268)
- 婴儿 (265)
- 水瓶座 (260)
- 射手座 (239)
- 不完美妈妈 (173)
- 跳槽那些事儿 (168)
- baby (140)
- 女婴 (132)
- 生肖 (129)
- 女儿 (129)
- 民警 (127)
- 狮子 (105)
- NBA (101)
- 家长 (97)
- 怀孕 (95)
- 儿童 (93)
- 交警 (89)
- 孕妇 (77)
- 儿子 (75)
- Angelababy (74)
- 父母 (74)
- 幼儿园 (73)
- 医院 (69)
- 童车 (66)
- 女子 (60)
- 郑州 (58)