首页 > 医疗资讯/ 正文
CAR-T耐药机制
嵌合抗原受体 (CAR)-T 细胞疗法最近成为治疗化疗难治性或复发性血癌患者的有力治疗方法,包括急性淋巴细胞白血病、弥漫性大B 细胞淋巴瘤、滤泡性淋巴瘤、套细胞淋巴瘤和多发性骨髓瘤。尽管如此,大多数患者还是会对 CAR-T 细胞疗法产生耐药性。
《Nature Reviews Drug Discovery》日发表综述,作者通过分析 CAR-T 细胞功能障碍、肿瘤固有耐药性和免疫抑制性肿瘤微环境,总结了 CAR-T 细胞免疫治疗的耐药机制;还讨论了目前克服多种耐药机制的研究策略,包括优化 CAR 设计、改善体内 T 细胞功能和持久性、调节免疫抑制性肿瘤微环境和协同组合策略。
现翻译全文供参考,因内容较多分为上下两篇予以分享,上篇主要是CAR-T细胞功能障碍导致的CAR-T耐药及克服策略,下篇聚焦于肿瘤固有耐药及克服抗原阴性逃逸策略、免疫抑制性肿瘤微环境及克服策略和展望。

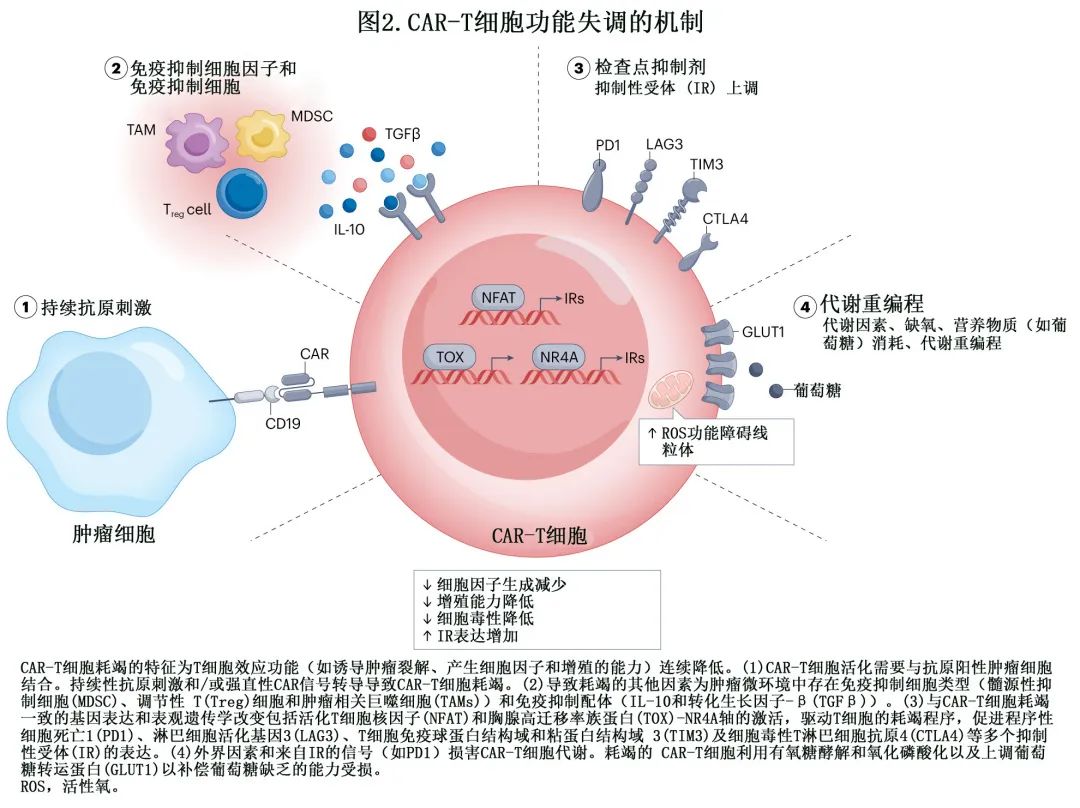
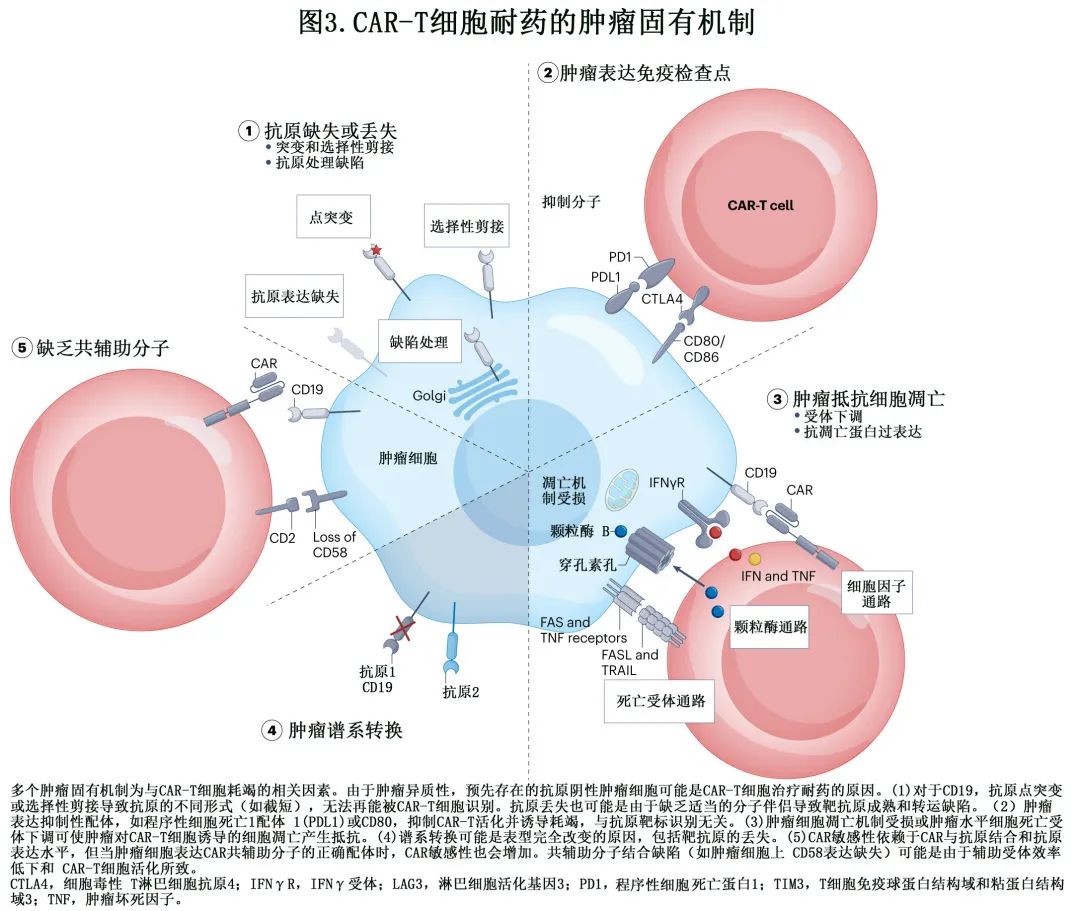
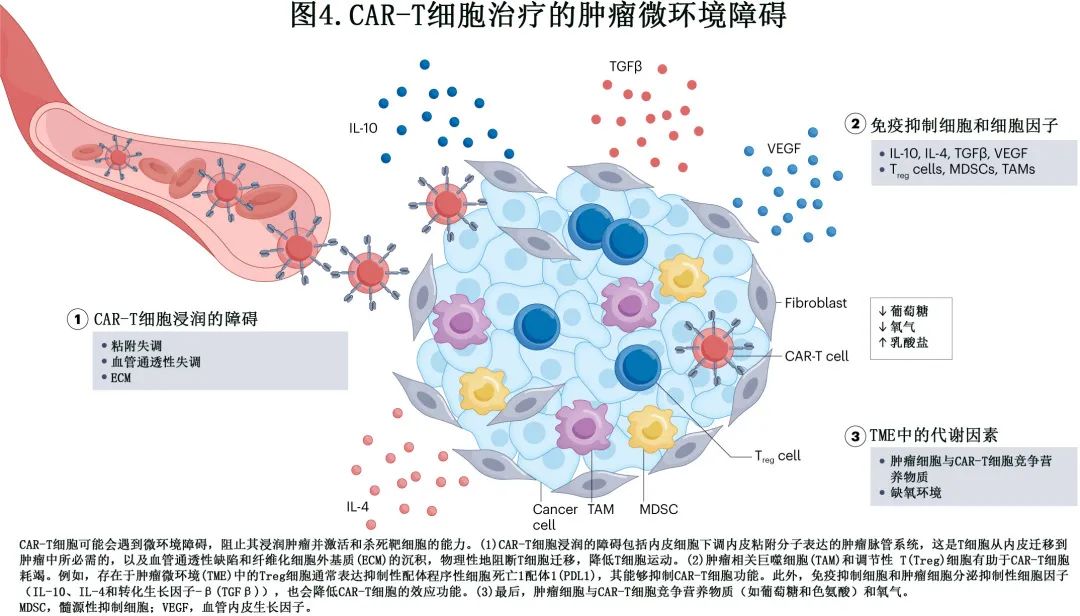
目前已经确定了CAR-T 细胞免疫治疗的耐药多种机制 (图1),即 CAR-T 细胞功能障碍(图2),肿瘤固有耐药机制 (图3)和免疫抑制性肿瘤微环境 (TME)(图4)。必须深入研究这些机制,以制定更有效的策略,实现患者的持久缓解。
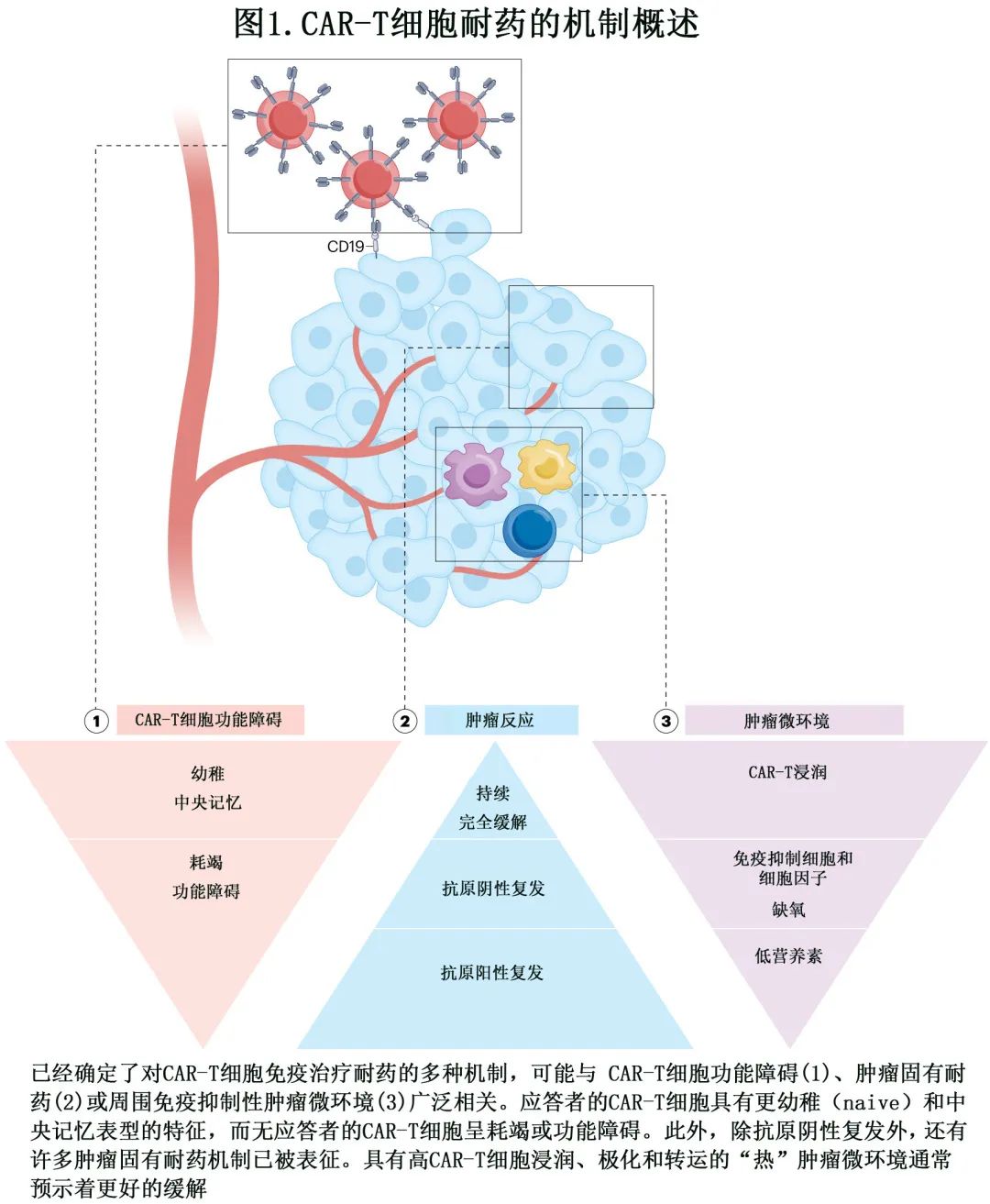
CAR-T细胞临床耐药
CAR-T细胞治疗后的原发性和继发性耐药特征因产品和疾病而异。在DLBCL注册研究(JULIET和 ZUMA-1)中,完全缓解率介于39%-58%。每种产品的注册研究在设计和患者人群上均存在差异,无法进行直接比较。然而,值得注意的是,在42%接受 axicabtagene ciloleucel(axi-cel) 治疗的患者、61%接受 tisagenlecleucel(tisa-cel) 治疗的患者和47%接受lisocabtagene maraleucel (liso-cel) 治疗的患者中观察到原发性耐药。这些结果证明,总的来说,淋巴瘤对 CD19的 CAR-T 细胞的原发性耐药发生于大约半数治疗患者中。
而B-ALL似乎对 CAR-T 细胞治疗的反应更强,ELIANA研究中81%的 B-ALL 儿童患者使用 tisa-cel 后达到完全缓解,ZUMA-3研究中 brexucabtagene autoleucel 的成人患者完全缓解率为53–60%。在 ZUMA-2 研究中,接受 brexucabtagene autoleucel 治疗的套细胞淋巴瘤患者中68%达到完全缓解,而在 ZUMA-5 研究中接受 axi-cel 治疗的滤泡性淋巴瘤患者的完全缓解率为74%。最后,在多发性骨髓瘤中,两种获批的 BCMA 靶向产品(idecabtagene vicleucel和ciltacabtagene autoleucel)的完全缓解率为67-79%。在更大型的真实世界研究中,axi-cel和 tisa-cel 报告的完全缓解率分别为53-65%和40-42%,与 ZUMA-1 和 JULIET 试验的结果相当。因此存在降低原发性耐药发生率的显著需求,尤其是在 DLBCL 和成人 B-ALL 中。
患者在达到完全缓解后复发时也可观察到继发性耐药,且复发时间因治疗的疾病和使用的产品而异。在中位随访40个月时,接受 tisa-cel 治疗 DLBCL 的患者的总生存期(OS)为11.1个月,中位无进展生存期 (PFS) 为2.9个月。ZUMA-1 研究的 axi-cel 随访分析(中位随访63.1个月)显示,5年OS率为43%,中位 PFS 为5.9个月。对于liso-cel,中位随访12.3个月,未达到中位OS。Tisa-cel治疗B-ALL的更新结果显示24个月无复发生存率为62%,18个月总生存率为70%。相同环境下的真实结果报告了相当的结果,总生存率为77.2%(中位随访时间为13.4个月)。ZUMA-2 研究中的套细胞淋巴瘤患者的缓解持续时间、PFS和OS分别为28.2、25.8和46.6个月。对于多发性骨髓瘤,idecabtagene vicleucel和 ciltacabtagene autoleucel 的2年随访获得的中位缓解持续时间分别为11.1和21.8个月(中位 PFS 为9.8个月,24个月 PFS 为54.9-60.5%)。
确定驱动原发性耐药和继发性耐药(或复发)的决定因素、机制和预后因素是一个非常活跃的研究领域。多项研究评估了在患者中常规评价的临床和实验室参数,并与较差结局相关。例如,较低的疾病负荷与较低的毒性和疗效增加相关,且这一原则适用于多种治疗方式和肿瘤疾病类型。此外,血液中 CAR 阳性 T 细胞达到峰值或曲线下面积水平时 CAR-T 细胞数量较多与缓解持久性较长相关。此外,在 tisa-cel 和 axi-cel 治疗 DLBCL 时,疾病复发的风险因素包括体能状态较差(美国东部肿瘤协作组[ECOG]≥2)、疾病分期较高、结外部位≥2个、乳酸脱氢酶 (LDH) 和/或 C-反应蛋白 (CRP) 实验室参数升高和国际预后指数(IPI)较高。治疗前较高的铁蛋白水平与较低的 CAR-T 细胞扩增相关。此外,较低的清淋前 LDH 水平以及较多数量的血小板是与较好的无事件生存期(EFS)相关的独立因素。需要“桥接化疗”(在血液成分单采和清淋化疗之间给予抗癌治疗,目的是防止在此期间疾病进展)的活动性疾病患者的总生存期更差。因此,尽管 CAR-T 细胞疗法在血液恶性肿瘤中取得了整体成功,但原发性和继发性耐药(尤其是后者)仍是重要的临床问题。
接下来的章节探索了对 CAR-T 细胞耐药的生物学机制,分为 T 细胞相关机制(如 CAR-T 细胞制造失败或 T 细胞耗竭)(图2)、肿瘤内在机制(如靶抗原表达缺失、表达免疫检查点分子或对细胞凋亡抵抗)(图3) 和免疫抑制性 TME 相关机制(如 CAR-T 细胞浸润的障碍、存在免疫抑制性细胞和细胞因子,或缺乏营养物质,或缺氧)(图4)。
CAR-T细胞功能障碍
T 细胞分化在 CAR-T 细胞持续存在中的作用,是理解 CAR-T 细胞治疗长期有效性的关键。T 细胞分化是指 T 细胞从幼稚状态转变为具有明显功能特征的效应或记忆状态的过程;在 CAR-T 细胞治疗中,CAR-T细胞在患者体内的持续存在与其能够在较长时间内维持其靶向癌细胞的效应功能密切相关。分化程度较低的 T 细胞亚群,如中央记忆 T 细胞和干细胞记忆 T 细胞,往往比分化程度较高的效应记忆 T 细胞和终末分化的效应细胞具有更优的持久性。中央记忆 T 细胞和干细胞记忆 T 细胞亚群具有自我更新能力,在重新遇到抗原时可以增殖,从而随着时间的推移维持 CAR-T 细胞群。相比之下,高度分化的亚群(如终末分化的效应细胞)的存在,由于其有限的增殖潜能和对耗竭或凋亡的易感性增加,可导致 CAR-T 细胞持久性下降。功能障碍的 T 细胞可能由多种原因引起,包括长期抗原刺激、既往淋巴毒性治疗、免疫缺陷或其他未确定因素。功能障碍的 T 细胞可以耗竭或衰老,并表现出较低的扩增能力、细胞毒性功能降低和较高的抑制性受体表达。减少耗竭和衰老的方法代表着一种增强 CAR-T 细胞功能的策略,已有多种方法正在研究中。表1提供了评估克服 CAR-T 细胞功能障碍新策略的临床试验概述,特别是关于阻断程序性细胞死亡 1(PD1) 及其配体,程序性细胞死亡1配体1(PDL1);用免疫球蛋白和 ITIM 结构域 (TIGIT) 阻断 PD1 和 T 细胞免疫受体的研究;CAR-T 细胞通过 BTK 依赖性和非依赖性机制与信号转导抑制相结合;使用自然杀伤 (NK) 细胞代替 T 细胞。

耗竭的 T 细胞最初在慢性病毒感染中报道,其特征是多种抑制性受体的共表达,包括PD1、细胞毒性 T 淋巴细胞抗原4(CTLA4)、T细胞免疫球蛋白结构域和粘蛋白结构域3(TIM3)、淋巴细胞活化基因3(LAG3)、TIGIT和其他。抑制性受体的表达数量和丰度与 T 细胞功能障碍的严重程度相关。此外,耗竭的 T 细胞表现为细胞因子(IFNγ、IL-2和肿瘤坏死因子[TNF])分泌受损和细胞毒性有限。耗竭的 T 细胞还表达与效应细胞和记忆细胞相关的转录因子,如活化 T 细胞核因子 (NFAT) 和下游效应因子NR4A、核胸腺高迁移率族盒蛋白 (TOX)、T-bet或eomesodermin,并采用独特的表观遗传景观,导致诱导耗竭程序,防止 T 细胞过度刺激并降低 T 细胞对肿瘤的反应。
具体而言,在 CAR-T细胞治疗中,临床前证据表明,从携带CD19+ B16-OVA黑色素瘤异异基因移植物的小鼠模型中分离的耗竭的CD8+ CAR-T细胞和肿瘤浸润淋巴细胞显示 PD1和 TIM3过表达,且NR4A和 NFAT转录因子的共有结合基序呈相似富集。在慢性淋巴细胞白血病(CLL)患者中,tisa-cel输注后的转录组学分析显示,与应答者的 T细胞相比,无应答者的 T细胞呈现与 T细胞耗竭、效应分化、糖酵解和凋亡相关的通路上调,且PD1、LAG3和 TIM3的共表达在这些 CAR-T细胞上也上调。治疗应答的可能性与输注高剂量CD27+ PD1-CD8+ CAR-T细胞之间存在高度显著相关性。同样,持续缓解与单采时记忆样 T细胞(CD27+CD45RO-CD8+)的频率较高和 IL-6和 STAT3相关基因标记的频率较高相关。在tisa-cel治疗DLBCL和转化滤泡性淋巴瘤的 JULIET研究中,PD1-PDL1相互作用评分最高和LAG3+ T细胞比例高的患者对 tisa-cel无应答或不久后复发。一项多中心回顾性研究报告,在76例 B细胞淋巴瘤(BCL)患者中,所有3例PDL1+肿瘤患者均对 axi-cel治疗原发性耐药。此外,最近的研究表明PDL1+ DLBCL对CD19 CAR-T细胞治疗难治,但 PD-1抑制剂帕博利珠单抗给药可导致 CAR-T细胞扩增和肿瘤消退。
自体 CAR-T 细胞治疗的另一个挑战在于 T 细胞直接来自患者,而血液恶性肿瘤患者(尤其是接受过多线细胞毒化疗的患者)具有更多的衰老 T 细胞和更少的记忆样CD8+ 细胞,而这些细胞与 CAR-T 治疗后的持久缓解相关。因此,输注时 T 细胞的状态可影响 CAR-T 细胞治疗的反应,输注前监测 CAR-T 细胞多功能性已证明是预测临床反应的关键属性。对 NHL 患者输注前 axi-cel 产品的单细胞分析表明,CAR产品含有能够部署多种免疫程序(细胞因子和趋化因子)的多功能 T 细胞亚群。应用于输注前 axi-cel 产品的预先规定的 T 细胞多功能性强度指数(polyfunctionality strength index)与临床反应显著相关。结合 CAR-T 细胞扩增或治疗前血清IL-15水平,多功能性强度指数可赋予额外的意义(NCT00924326)。
其他的工作也已经显示出在制备过程中使用 T 细胞预选具有希望,因为它可去除潜在的功能障碍的 T 细胞。预选的初始/干细胞记忆 T 细胞可提供更高的抗肿瘤活性,并对 CAR-T 细胞诱导的毒性表现出较低的易感性。
克服CAR-T细胞功能障碍的策略
优化CAR-T细胞产品
之前的许多综述详细讨论了各种 CAR 结构设计,特别是结合剂、铰链结构域和共刺激结构域,可增加 CAR-T 细胞抗原识别、信号传导和持久性。本文重点关注 CAR 设计以外的其他方面,而这些方面可以影响 CAR-T 细胞产品。
在大多数血液恶性肿瘤患者中,由于存在肿瘤和既往细胞毒性治疗,T细胞耗竭并终末分化,即使进行离体活化和工程,来源于这些患者的 CAR-T 细胞相对于来源于年轻、健康供者的 T 细胞仍表现稍差。降低功能障碍T细胞分数的潜在策略为,从分化程度较低的骨髓浸润 T 细胞开始,而非循环外周血 T 细胞,但需要更具侵入性的操作。或者使用在病程早期采集的 T 细胞(暴露于淋巴毒性治疗较少),它可以保留比复发后采集的T细胞更好的健康度(fitness)和增殖能力。然而尽管如此,任何阶段存在的疾病都可能影响 T 细胞功能,并存在于最终产品中。在 ZUMA-12研究(NCT03761056) 中检测了 axi-cel 作为高危大 BCL(LBCL) 患者一线治疗的疗效和安全性,发现与 ZUMA-1 相比,更高比例的幼稚(naive)样 T 细胞 (CCR7+CD45RA+) 与 CAR-T 细胞的更大扩增相关,表明将 CAR-T 细胞纳入更早线的细胞系中可增强 T 细胞功能和临床疗效。
缩短 CAR-T 细胞的培养期也有利于制备细胞毒性更强、分化程度更低的细胞产品。利用慢病毒载体进入并整合到非分裂细胞基因组中的独特能力,可以在一天内生成高功能的 CAR-T 细胞。尽量减少离体操作可保留较少耗竭的 T 细胞,也可能对疾病快速进展的患者特别有益,否则这些患者可能无法接受该治疗。当然,由于放行前完成测试所需的时间,仍需确定交付患者的时间是否会显著缩短。
为了更好地理解导致耗竭的机制,Lynn等人最近通过在 T 细胞中引入tonic信号 CAR(HA-28z CAR) 建立了细胞耗竭模型。与基因表达分析配对的染色质显示转录因子、激活蛋白 1(AP-1) 基序的可及性增加,IRF4和 BATF 优先结合JUN,因为这些转录因子竞争相同的位点。JUN的强制过表达可通过促进 AP-1 靶基因的直接转录激活以及通过间接破坏驱动耗竭相关基因表达的免疫调节AP-1–IRF转录复合体来阻止耗竭。这些结果显示了表观遗传学和转录重编程如何逆转 CAR-T 细胞耗竭。
细胞因子可刺激 CAR-T 细胞增殖超过耗竭的正平衡。在表达 IL-7 受体与 CAR 组成型活性形式或在 CAR 结构中插入 IL-15 受体的 CAR-T 细胞可发现到抗肿瘤活性增加。额外的转基因也可以掺入到相同的载体构建体中,以增强 CAR-T 细胞持久性,如 BCL-2 家族成员。避免细胞死亡也可以通过显性负相 FAS 过表达或FAS–41BB开关受体来完成。CAR-T 细胞中全基因组 CRISPR 筛选新出现的数据也表明,删除其他基因(如RASA2)可能会增强 CAR-T 细胞制备、持久性或功能。该领域的一个挑战在于,可以在临床前模型中假设和测试的潜在编辑、转基因和敲除的数量远远超过可能为患者临床开发的潜在产品的数量。
阻断免疫检查点
抑制PD1可能克服PDL1+ 肿瘤免疫抑制,从而显著增强人 CAR-T 细胞对血液学和潜在实体瘤的抗癌活性。通过使用抗体阻断 PD1 可通过恢复抗癌 T 细胞免疫,在各种癌症类型中均表现出显著的临床疗效;然而同时也观察到免疫相关不良事件,尤其是自身免疫。
一项研究纳入14例既往接受过多线治疗的难治性 B-ALL 儿童患者,聚焦于在 CD19 CAR-T 细胞治疗的基础上加用 PD1抑制剂(其中13例接受帕博利珠单抗,1例接受纳武利尤单抗),结果 CAR-T 细胞的持久性得到改善,50%(14例患者中的7例)维持完全或部分缓解。此外,接受 PD1 抑制剂治疗的患者中有3例发生复发性 B 细胞再生障碍性贫血,提示 CAR-T 细胞功能持续存在。
通过基因编辑靶向调节 CAR-T 细胞产物中的PD1,具有出保护 CAR-T 细胞免受 PD1 诱导的耗竭的优势,同时保留旁观(bystander) T 细胞,从而降低自身免疫毒性的风险。阻断该通路的另一种策略是通过“装甲(armoured’)”CAR-T细胞递送 PD1 抑制剂,该细胞可分泌免疫检查点阻断单链可变片段 (scFv)。该策略在PDL1+血液恶性肿瘤的临床前同基因和异基因小鼠模型中均显示出极好的结果。其他抑制 PD1 的巧妙方法,如引入显性失活(dominant)PD1胞外域或新型 PD1-CD28 嵌合开关受体(chimeric switch receptor),也已在实体瘤免疫治疗中开发。PD1–CD28嵌合开关受体包含与细胞内共刺激分子 CD28 融合的 PD1 胞外结构域,具有两个目的:结合其配体 PDL1 和传递激活信号而非抑制信号。与第二代 CD19 CAR-T 细胞相比,携带该构建体的 T 细胞在体外和体内均对PDL1+淋巴瘤细胞表现出优越活性。值得注意的是,采用这种方法治疗的1例患者获得持久临床缓解且无显著毒性作用。
最后,Lee等人最近使用了一种慢病毒二合一CAR 构建体,两个检查点受体被整合到 CAR 载体中的双短发夹 RNA 盒同时下调。利用该系统,作者评估了 CD19 CAR-T 细胞与四种检查点组合——PD1–TIM3、PD1–LAG3、PD1–CTLA4和PD1–TIGIT,发现 PD1 和 TIGIT 的下调具有独特的协同抗肿瘤作用。在研究中,PD1的下调可增强CAR-T 细胞的短期效应功能,而 TIGIT 的下调负责维持低分化/耗竭状态,从而提供潜在的协同作用。目前正在复发性或难治性 LBCL 成人患者中评价PD1–TIGIT下调的 CD19 CAR-T 细胞的疗效和安全性 (NCT04836507)。许多进行中的临床试验也评估了免疫检查点阻断联合 CD19 CAR-T 细胞(如NCT04163302)的效果。检查点抑制的最佳时机和组合仍有待确定,且可能出乎意料;例如在 T 细胞受体 (TCR) 转导 T 细胞的临床研究中,PD1的基因敲除似乎赋予缺点而非增加持久性。
联合小分子
减少 T 细胞功能障碍的一个可能策略是将 CAR-T 细胞与可利用 T 细胞生物学增强 T 细胞活性的小分子结合。例如,酪氨酸激酶抑制剂(如伊布替尼)与 CD19 CAR-T 细胞联用时可增强抗肿瘤活性。伊布替尼可抑制 B 细胞受体 (BCR) 下游激酶 Bruton 酪氨酸激酶 (BTK),也可靶向 IL-2 诱导的 T 细胞激酶 (ITK),ITK是参与调节磷脂酶Cγ的 T 细胞中的一种酪氨酸激酶,位于 TCR 信号转导的下游,对辅助性 T 细胞2型效应应答至关重要。ITK 的缺失或抑制会驱动更多的辅助性 T 细胞1型反应,在针对 CLL 患者的临床前和临床研究中,伊布替尼可增强 CAR-T 细胞活性、增殖、细胞因子的产生和 CAR-T 细胞的植入。此外,T 细胞采集时既往接受伊布替尼治疗≥6个月的 CLL 患者显示体外和体内 CAR-T 细胞扩增改善,与健康年轻供者的 T 细胞扩增相当,这一发现与临床缓解呈正相关。此外,如果同时给药,伊布替尼暴露可改善耐药 ALL 和 CLL 异基因移植小鼠模型中的 CAR-T 细胞植入、肿瘤清除和生存。套细胞淋巴瘤 CAR-T 细胞的临床前模型中也获得结果,该方法由多个小组转化为临床。Gill 等人在接受伊布替尼治疗>6个月但疾病持续的 CLL 患者中证实,自体 CAR-T 细胞(使用人源化结合剂靶向CD19)生产期间 T 细胞增殖更优。这项前瞻性单中心研究 (NCT02640209)报告了18例患者至少随访15个月的结果;值得注意的是,其中3例患者既往接受过携带鼠 scFv 的 CD19 CAR-T 细胞治疗但疾病进展。12个月时,72%的患者无微小残留病,48个月时的估计OS和 PFS 分别为84%和70%。细胞因子释放综合征 (CRS) 常见,但为轻-中度且不需要治疗,此外观察到体外 T 细胞扩增改善。该结果与纪念梅隆凯瑟癌症中心的临床试验数据一致,该研究显示,在 T 细胞采集和/或 CAR-T 细胞给药时接受伊布替尼治疗的16例 CLL 患者中,有5例出现显著的 T 细胞增殖。最后,Gauthier等人表明,在白细胞单采前至少2周开始每天420mg,并在 CAR-T 细胞输注后持续至少3个月的情况下,伊布替尼的耐受性良好;与未接受伊布替尼治疗的患者相比,同时接受伊布替尼治疗的患者体内 CAR-T 细胞数量更多,CRS严重程度更低(中位 CRS 分级分别为1级和2级;P=0.04)。此外在接受伊布替尼治疗的患者中,通过多参数流式细胞术 (72%) 和免疫球蛋白重链测序 (85%) 测量,增殖指数更高、多功能 CAR-T 细胞(表达≥2种细胞因子)的百分比更高、骨髓中无微小残留病的患者比例更高。目前尚不清楚二代 BTK 抑制剂(对 BTK 更具有特异性且不靶向ITK)是否会获得相似的临床结果。
另一种酪氨酸激酶抑制剂达沙替尼已与 CAR-T 细胞治疗联合使用具有双重用途。达沙替尼可干扰淋巴细胞特异性蛋白酪氨酸激酶 (LCK),阻止 TCR 下游CD3ζ和 ZAP70 的磷酸化。该机制可通过破坏携带CD28-CD3ζ或4-1BB–CD3ζ活化模块的 CAR-T 结构中的下游信号级联,增加 CAR-T 细胞的安全性指数。在临床可达到的浓度 (100nM) 下,达沙替尼可关闭活化的 CAR-T 细胞,防止细胞毒性,而不损害其活力;且这种‘CAR-T暂停状态’是可逆的,停用达沙替尼可恢复 CAR-T 细胞溶解活性、细胞因子分泌和增殖。在植入工程化表达 GD2 的 Nalm6 白血病细胞的小鼠中,在 GD2 CAR-T 细胞 (GD2.28ζ) 治疗期间引入达沙替尼。FKBP)可促进 CAR 信号转导的一过性停止,减轻耗竭,促进体外扩增过程中的 T 细胞记忆,增强体内 CAR-T 细胞抗肿瘤功能。
PI3K–AKT通路在TCR、共刺激分子和细胞因子受体下游发挥至关重要的作用。PI3K–AKT的抑制剂可增强 T 细胞中央记忆表型、减少终末分化和增加增殖,因此人们探索了使用 AKT 抑制剂来延长 CAR-T 细胞持久性。临床前研究表明,在 CD19 CAR-T 细胞生产过程中 AKT 信号转导的抑制可导致 T 细胞在体外和体内具有优越的抗肿瘤活性;且在抗 BCMA CAR-T 细胞的生产过程中也有效。IL-2 与 PI3K 抑制剂 bb007 联合使用可导致 CAR-T 细胞产物 (bb21217) 富集于记忆样 T 细胞中,减少分化并改善扩增。这些数据表明,PI3K–AKT的治疗性调节可能是增强 CAR-T 细胞治疗抗肿瘤疗效的策略,一旦 AKT 抑制剂被批准用于临床就可以过渡到临床。
总体而言,小分子是增强 CAR-T 功能的重要工具,但剂量、时间表和长期效果需要在临床上进一步测试。
异基因T细胞
CAR-T 细胞可以使用来自健康供者的 T 细胞进行制备,其与患者来源的 T 细胞相比具有多个潜在优势。首先,健康供者可能提供功能更强的 T 细胞产物,避免 T 细胞功能障碍。此外可限制制备失败的风险(该问题发生于10-20%的 CAR-T 细胞批准研究中),并减少库存和可用的现成 CAR-T 细胞的输注时间。然而与任何异基因治疗(如干细胞移植)一样,移植物抗宿主病 (GVHD) 和 CAR-T 细胞排斥仍是需要克服的主要障碍。为了避免这些并发症,不同的策略正在开发中。基因编辑、选择特定亚群的 T 细胞(γδT细胞或病毒特异性 T 细胞)或健康供者的 NK 细胞可逃避GVHD。此外,可以敲除TCRα链恒定基因 (TRAC) 的异基因αβT细胞。也可以使用额外的基因编辑或隐形(stealth)策略来避免宿主对治疗产品的排斥,例如利用CRISPR–Cas9或其他编辑技术修饰与主要组织相容性 I 类复合体 (MHC-I) 相关的位点。然而,尽管有一些有前景的初步数据,但迄今为止其总体结果尚未达到与自体产品相同的疗效水平。
靶向 CD19 的基因编辑的异基因 CAR-T 细胞,如‘UCART19’,已经显示出一定的疗效。7例儿童和14例成人作为 UCART19 的一部分接受治疗,21例患者中的14例 (67%) 在输注后28天达到完全缓解或完全缓解伴血液学不完全恢复;但未接受阿仑单抗的患者(4例患者)未显示 UCART19 扩增。3例 (14%)发生3-4级CRS,其他不良事件包括2例患者 (10%) 的 I 级急性皮肤移植物疾病和6例患者 (32%) 的长期 IV 级血细胞减少。记录了2例治疗相关死亡:1例患者发生中性粒细胞减少性败血症伴CRS,另1例患者发生肺出血伴持续性血细胞减少。
因此,尽管在很大程度上可以预防 GVHD 等异基因反应的发生,但排斥 allo-CAR-T 仍是实现持久缓解的主要障碍;但UCART19可作为一种潜在的桥接移植选择。
避免异基因治疗性细胞产品的排斥反应需要同时阻止 T 细胞和 NK 细胞反应,以消除外源细胞。一种合成受体,同种免疫防御受体 (ADR) 已被开发出来,可选择性识别活化 T 细胞中表达的4-1BB。表达 ADR 的 T 细胞可靶向异基因反应性(alloreactive)淋巴细胞,同时保留沉寂(quiescent)淋巴细胞,从而抵抗排斥反应。设计用于表达 ADR 的 CAR-T 细胞在血液瘤和实体瘤体内模型中均表现出持续的肿瘤根除,因此提供了一种产生抗排斥异基因 T 细胞产品的现成方法。其他研究小组旨在使健康供者 T 细胞对淋巴细胞耗竭剂耐药,因为强化清淋可能使异基因 CAR-T 细胞在宿主免疫恢复前增加扩增和抗肿瘤活性。脐带血来源的 T 细胞可能作为除健康供者 T 细胞以外的另一种 T 细胞来源,在接受异基因干细胞移植的患者中,由于释放的促炎细胞因子水平较低可降低 GVHD 频率。其他选择,如来源于诱导多能干细胞的免疫效应细胞也处于早期发展阶段。
非T细胞免疫效应细胞
在T细胞疗法成功的基础上,过继细胞治疗策略已经开始包括其他免疫效应细胞或 T 细胞亚群,如 NK 细胞、巨噬细胞、γδT细胞和恒定自然杀伤 T(iNKT) 细胞。
NK细胞。NK 细胞是细胞毒性细胞,在针对病毒感染或细菌感染和损伤细胞的先天性免疫应答中发挥重要作用。由于其固有特性,它们可提供作为异基因细胞免疫治疗的潜力,因为它们不识别同种异体抗原,也不引起GVHD。然而由于 MHC 的表达,它们仍然容易受到宿主免疫系统的异基因排斥反应的影响。NK 细胞的活化受一组不同的跨膜受体的调节,包括活化、抑制、细胞因子和趋化因子受体。MHC 下调是肿瘤细胞的共同特征,可为 NK 细胞提供一种通过“缺失-自我”机制杀伤的活化机制。此外,NK细胞拥有激活受体,如 NKG2D 和激活杀伤免疫球蛋白样受体 (KIR),能够识别癌细胞优先表达的“诱导自身”配体。NK 细胞的另一个潜在优势依赖于其杀伤机制,可通过 CAR 转导增强,CAR-NK 细胞可通过 CAR 依赖性或非 CAR 依赖性机制消除靶癌细胞。大多数用于 NK 细胞的 CAR 构建体与 CAR-T 细胞中使用的结构相同,具有CD3ζ胞内结构域和 4-1BB 共刺激结构域,以改善 CAR-NK 活化和细胞因子生成(如IFNγ、粒细胞-巨噬细胞集落刺激因子 (GM-CSF))。其他结构使用 NK 细胞特异性共刺激结构域(DAP12和2B4)设计,在细胞毒性和体内抗肿瘤作用方面可能优于4-1BB。
CAR-NK 细胞的理论优势让人们产生了相当大的热情,许多临床研究正在进行中。目前至少已经计划或激活了至少32项 CAR-NK 细胞的临床试验,其中11项针对急性白血病,10项针对淋巴瘤,1项针对多发性骨髓瘤的产品;靶抗原为CD19、CD22、CD19/CD22、CD33、BCMA和CD7。
来源于成人外周血单个核细胞的 NK 细胞很难转导,但 NK 细胞更适合基因工程,可以从脐带血、成人骨髓CD34+造血干细胞或诱导性多能干细胞中获得。在 NHL 或 CLL 患者中开展的 I/II 期临床试验显示,脐带血 NK 细胞转导抗CD19 CAR 并经基因工程改造表达 IL-15 可改善持久性(在发生不良事件但未触发细胞凋亡的情况下,纳入嘧啶诱导的半胱天冬酶9自杀基因作为触发细胞凋亡的安全性指标),患者完全缓解并持续缓解;11例患者中8例患者对治疗有反应,7例患者完全缓解,1例患者 Richter 转化成分缓解但 CLL 持续。有趣的是,NK细胞在输注后持续约2周,从而具有在靶向脱靶效应情况下限制细胞毒性的潜在优势;然而需要多次输注 CAR-NK 细胞才能实现肿瘤清除。值得注意的是,本试验中的患者数量有限,大多数缓解患者在 CAR-NK 细胞后接受了额外的治疗,并且没有长期随访。
现在有多种分化方案可用于从可诱导多能干细胞中产生 NK 细胞,这是 NK 细胞的可再生来源,可作为克隆和基因组编辑的平台。它们可能导致生产成本降低,因为可以在不同的时间冷冻保存并给予患者。
尽管 CAR-NK 细胞治疗相对于 CAR-T 细胞治疗具有一定优势,但也存在重要的局限性。NK 细胞是有效的细胞毒性细胞,但也可以在 TME 功能耗竭,主要是因为缺氧。尽管如此,缺氧可以作为构建 CAR-NK 细胞时的优势。Juillerat 等人证明在 CAR 构建体中掺入一个低氧诱导因子1α (HIF1α) 结构域可以生成一个系统,在这个系统中 CAR 的表达受到 TME 中低氧的刺激。此外,CRISPR–Cas9可以用来改变参与 NK 细胞耗竭或功能的通路,或对抗转化生长因子-β (TGFβ) 免疫抑制。
巨噬细胞。巨噬细胞是先天性免疫系统的细胞组分,可以转运到肿瘤细胞,调节 TME 并呈递各种抗原。克服免疫抑制性 TME 和负性免疫调节信号是 CAR-T 细胞面临的主要挑战之一,因此CAR-巨噬细胞已成为一种可能的替代工具,尤其是针对实体瘤,因其可浸润 TME 并调节免疫应答。产生基于巨噬细胞的免疫治疗时的一个关键挑战在于使其促炎作用成为可能,设计用于识别具有 CD8 跨膜和CD3ζ胞内结构域的CD19、HER2或间皮素的 CAR 巨噬细胞可有效地将巨噬细胞重定向至 M1 表型,刺激抗原依赖性吞噬作用、细胞因子释放和抗肿瘤活性。Morrissey 等人改造了吞噬作用的 CAR 家族,使巨噬细胞吞噬特异性抗原包被颗粒(CD19和CD22),并检测了一组胞内结构域。但由于巨噬细胞进化为检测和反应外源性核酸,并表现出有限的增殖能力,CAR结构的递送具有挑战性。Klichinsky 等人使用巧妙的复制缺陷型嵌合腺病毒载体 Ad5f35 递送CAR,该系统转导巨噬细胞具有高效率和可重复性,并导致 M1 分化,从而产生促炎性 TME 并与 CAR 活性协同作用。
目前多项评估 CAR-巨噬细胞治疗实体瘤的临床试验正在进行中 (NCT04660929)。
参考文献
Ruella M, et al. Mechanisms of resistance to chimeric antigen receptor-T cells in haematological malignancies.Nat Rev Drug Discov . 2023 Oct 31. doi: 10.1038/s41573-023-00807-1.
- 搜索
-
- 1000℃Nutrients:真实世界数据,纤维肌痛患者的饮食与运动自适应规律
- 1000℃D-二聚体升高诊治与管理专家共识(2026)
- 1000℃专家论坛|文良志:门静脉血栓的诊断和治疗
- 1000℃首例儿童NF2驱动型胸膜间皮瘤,多方法学检测锁定NF2双等位基因失活和14/22号染色体缺失,提示与成人胸膜间皮瘤不同
- 1000℃打破误区:干扰素追求CHB功能性治愈,HBsAg为何“不降反增”?
- 1000℃迷惑性极强的肝内病灶!影像表现疑点重重,最终病理竟查出两种不同肝脏恶性肿瘤
- 1000℃指南共识|原发性肝癌分子靶向药物相关蛋白尿中西医结合诊疗专家共识
- 1000℃Diabetologia:意大利北部社区 1~100 岁人群胰岛自身抗体与乳糜泻 TGA-IgA 的年龄分布及检测方法学验证
- 精J Child Psychol Psychiatry:12种罕见神经发育障碍儿童沟通能力谱系
- 精研究发现:爱吃辣的人,心血管病和癌症死亡风险都会显著降低
- 精Nursing in Critical Care:别再指责护士了!ICU 里被遗漏的护理,根源在系统而非个人
- 精Acta Obstet Gynecol Scand:罕见病女性的妊娠并发症与母婴结局,一项单中心434种罕见病的回顾性队列研究
- 精【爱儿小醉】儿科患者术前对流层臭氧暴露与围手术期呼吸系统不良事件之间的关系:一项单中心回顾性队列研究
- 精eBioMedicine:牙龈下微生物组与脑健康存在连续关联梯度,牙周炎或成认知衰退可干预靶点
- 精军事医学研究院《自然·通讯》:自适应IrPtCu纳米酶水凝胶实现耐药菌感染伤口序贯治疗
- 精能够逆转萎缩性胃炎的两个中成药,该怎么选择?
- 荐同时性多发性原发性肺癌,左右病灶分别为EGFR和ALK阳性,考虑淋巴结肿大仅局限左肺门及血浆EGFR阳性,采用奥希替尼联合化疗
- 荐40岁女性同时罹患卵巢支持细胞-间质细胞瘤和透明细胞乳头状肾肿瘤,WES等基因检测竟为阴性
- 荐椎管内麻醉使用止血药突发气道痉挛的抢救流程解析
- 荐女子肝区无任何不适,影像提示复杂囊性病变,层层鉴别后锁定罕见胆管源性囊性肿瘤
- 荐“绘”真报告 | 病理考虑为中枢神经细胞瘤,检出脑室外神经细胞瘤的特征性变异FGFR1-TACC1融合,辅助鉴别诊断
- 荐8岁女童出现男性化症状,竟是形似「性索-间质肿瘤」的卵巢「无性细胞瘤」所致,少见KRAS/CDK4共扩增或与侵袭性有关
- 荐17例病例分析揭示常见于中年人的色素性室管膜瘤临床特征与预后,分子检测可助力临床精准诊疗
- 荐Lancet Oncol:结直肠癌腹膜转移,围手术期化疗并非必选项
- 标签列表
-
- 星座 (702)
- 孩子 (526)
- 恋爱 (505)
- 婴儿车 (390)
- 宝宝 (328)
- 狮子座 (313)
- 金牛座 (313)
- 摩羯座 (302)
- 白羊座 (301)
- 天蝎座 (294)
- 巨蟹座 (289)
- 双子座 (289)
- 处女座 (285)
- 天秤座 (276)
- 双鱼座 (268)
- 婴儿 (265)
- 水瓶座 (260)
- 射手座 (239)
- 不完美妈妈 (173)
- 跳槽那些事儿 (168)
- baby (140)
- 女婴 (132)
- 生肖 (129)
- 女儿 (129)
- 民警 (127)
- 狮子 (105)
- NBA (101)
- 家长 (97)
- 怀孕 (95)
- 儿童 (93)
- 交警 (89)
- 孕妇 (77)
- 儿子 (75)
- Angelababy (74)
- 父母 (74)
- 幼儿园 (73)
- 医院 (69)
- 童车 (66)
- 女子 (60)
- 郑州 (58)